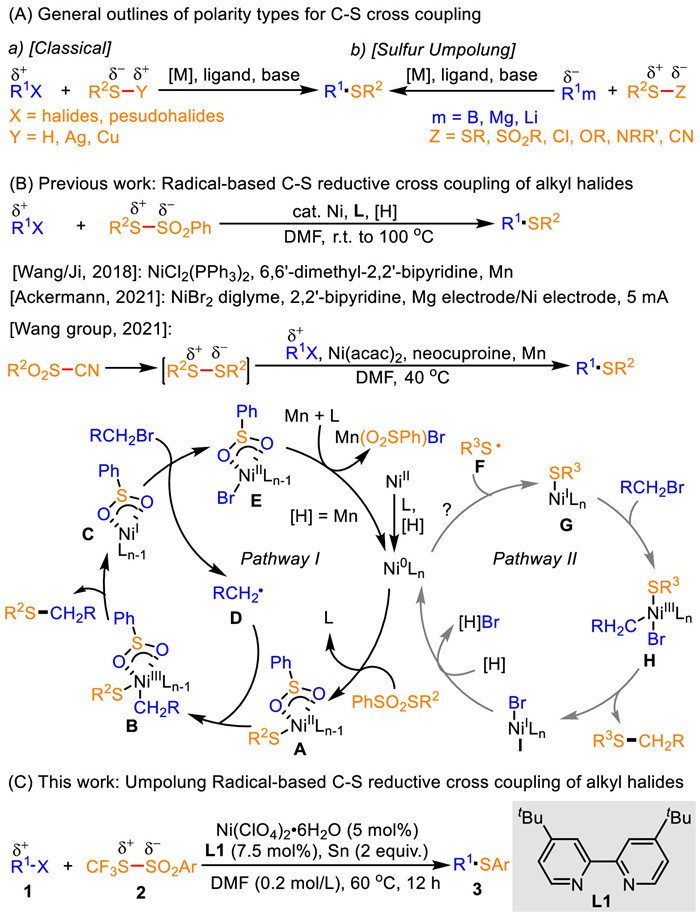
Scheme 1.
Synthesis of thioethers.
Nickel-catalyzed umpolung C–S radical reductive cross coupling of S-(trifluoromethyl)arylsulfonothioates with alkyl halides
Yu-Zhong Yang , Gui-Fen Lv , Ming Hu , Yang Li , Jin-Heng Li

Organosulfur compounds, including thioethers, are core moieties encountered in the structure of various drugs, natural products, herbicides, ligands and functional materials, as well as valuable synthetic building blocks and latent functional groups that can be modified to assemble complex target molecules in synthesis [1-16]. As a result, significantly ongoing efforts have been devoted to the development and expansion of methods for catalytically forging highly valuable and functionality diverse thioether scaffolds in synthetic and medicinal chemistry [8-16]. Conventional methodologies to straightforward access thioethers involve transition-metal-catalyzed C—S cross coupling reaction [8-16], which is dominated by two different modes of reactivity, including a classical-polarity method using the thiol functionality as a nucleophile (Scheme 1A-a) [17-32] and an umpolung approach employing the sulfur-based reactant as an electrophile (RSX, X = SR, SO2R, Cl, OR, NRR’ or CN; Scheme 1A-b) [33-42]. While these polarity modes of catalytic C—S cross couplings of aryl halides or aryl organometallic reagents (such as arylboronic acids, arylmagnesiums and aryllithiums) with the thiolation reagents for producing aryl-tethered thioethers by incorporation of an aryl group onto a sulfur atom to form a C(sp2)-S bond have been well established and widely exploited [8-42], analogous versions to access alkyl-tethered thioethers via introduction of an alkyl group onto the sulfur atom to construct the C(sp3)-S bond have been less extensively studied [8-16,29], probably due to tendence to the facile side reactions (such as β-hydrogen elimination) under strong alkaline and elevated temperature conditions. Furthermore, the vast majority of these reported protocols suffer from the use of highly toxic, air sensitive, odor disagreeable thiols and their oxidized derivatives, as well as only few commercially available alkyl thiols and alkyl disulfides, which significantly impede their widespread applications. Therefore, these challenges and the increasing importance of alkyl-tethered thioethers spur the synthetic chemists to develop mild, versatile strategies that (ⅰ) enable efficient incorporation of an alkyl group onto a sulfur atom to form the C(sp3)-S bond under base-free conditions; (ⅱ) accommodate broad functionalized substrates, especially including diverse alkyl halides and readily accessible, bench-stable, odourless thiolation reagents; and (ⅲ) are subject to facile late-stage modification of biologically relevant molecules.
To circumvent these issues, transition-metal-catalyzed C—S reductive cross coupling reaction of organohalides with electrophilic thiolation reagents has recently been developed as a promising alternative to the conventional polarity types with preformed nucleophiles (Scheme 1B) [22,43-57]. These approaches allow facile introduction of an electrophilic aryl or alkyl group onto the electrophilic sulfur atom to construct the sp2- and sp3-hybridized C—S bonds under mild and base-free conditions, and thus exclude side reactions, such as β-hydrogen elimination. However, only few approaches have been reported to allow catalytic C—S reductive cross couplings of unactivated alkyl halides with electrophilic thiolation reagents (e.g., disulfides and thiosulfonates) for producing alkyl-tethered thioethers. For example, the group of Wang/Ji has reported the first nickel-catalyzed C—S reductive cross coupling of unactivated alkyl bromides with thiosulfonates and Mn reductant [46-48], which is highlighted by the use of the simple, bench-stable and odorless thiosulfonates as the electrophilic thiolation reagents and by a plausible mechanism comprising an inner-sphere Ni0/Ⅱ/Ⅲ/Ⅰ/Ⅱ catalytic cycle directly engaged by the alkyl carbon-centered radicals from homolysis of alkyl halides. Later, this group developed a similar catalysis version to accomplish thiolation of alkyl bromides with arenesulfonyl cyanides as the electrophilic disulfide precursors for assembling alkyl aryl sulfides [50]. Very recently, the group of Ackermann reported an electroreductive nickel-catalyzed radical thiolation by cross-electrophile coupling of alkyl bromides with functionalized thiosulfonates through Mg cathodic reduction to give alkyl-tethered thioethers [51]. These methods rely on the generation of the alkyl carbon-centered radical D from alkyl halides reacted with the in situ formed NiⅠ intermediate C, which would sequentially execute single electron oxidation with the NiⅡ intermediate A to afford the NiⅢ intermediate B (Pathway Ⅰ; Scheme 1B) [45-50]. On the basis, we hypothesized that initially generating the sulfur-centered radical F, which are formed from homolysis of the electrophilic thiolation reagent components, would give rise to single electron oxidation to deliver the NiⅠ-SR intermediate G followed by oxidative addition with alkyl halides to produce the NiⅢ intermediate H (Pathway Ⅱ; Scheme 1B), which would: (ⅰ) provide new radical reductive cross-coupling tactics comprising the engagement of the reaction with the sulfur-centered radicals thus resulting in access to otherwise poorly accessible or unobtainable molecular frameworks; (ⅱ) expand the reactivity profile of Ni reductive catalysis; and (ⅲ) innovate and advance radical chemistry.
Herein, we report the first nickel-catalyzed DMF-mediated umpolung C—S radical reductive cross coupling between S-(trifluoromethyl)arylsulfonothioates and alkyl halides involving a sulfur-centered radical formation (Scheme 1C). This reaction is initiated by DMF, Ni(ClO4)2·6H2O, 4,4′-di-tert-butyl-2,2′-bipyridine L1 and Sn, and enables the formation of the C(sp3)-S bonds through umpolung transformations of S-(trifluoromethyl)arylsulfonothioates and sequential catalytic reductive cross coupling with alkyl halides.
To determine the role of arylesulfonothioates 2 as the S-based functional group sources, the umpolung C—S radical reductive cross coupling of 3-phenylpropyl bromide 1a with PhSO2SCF3 2a was examined (Table 1). Screening various reaction parameters revealed that a combination of 5 mol% Ni(ClO4)2·6H2O, 7.5 mol% 4,4′-di-tert-butyl-2,2′-bipyridine L1 and 2 equiv. Sn in DMF (0.2 mol/L) at 60 ℃ for 12 h afforded the desired phenyl(3-phenylpropyl)sulfane 3aa in nearly quantitative yield with excellent chemoselectivity (entry 1). Unlike the previously reported results acted as the SCF3 (often) or PhSO2 source [58-68], PhSO2SCF3 2a serves as the PhS source. Both Ni catalysts and Sn are necessary to make the reaction successful as leaving out each led to no desired reaction (entries 2 and 16), and a lower loading of Ni(ClO4)2·6H2O (2 mol%) decreased the yield (entry 3). Other Ni catalysts, including NiCl2, NiBr2, NiCl2·DME, NiCl2(PPh3)2 and NiCl2(Py)4, were highly active (entries 4–8), but all were less efficient than Ni(ClO4)2·6H2O. Optimization of the dinitrogen-based ligand effect indicated that these ligands L1-L6 served as promotors since omission of ligands the reaction could still run efficiently to tender 3aa in 86% yield (entries 9–15). Furthermore, ligands L2, L4-L6 could improve the reaction (entries 11 and 13–15), but 2,2′-bipyridine L3 was detrimental to the reaction outcome attributing to strong coordination with the Ni catalyst lowering its catalytic activity (entry 12). Using the same equivalent amount of Ni(ClO4)2·6H2O and L1 slightly diminished the yield (entry 10), suggesting that excess L1 assists complete reduction of Ni(ClO4)2·6H2O to the active Ni0 species avoiding consumption of Sn reductant. The yield raised from 84% to 95% with the increase of the Sn amount from 1.2 equiv. to 1.7 equiv. (entry 17). These observations indicate that the roles of Sn mainly include reduction of PhSO2SCF3 and regeneration of the active Ni(0) species. Notably, the reaction is sensitive to the reducing reagents as the other common reductants, such as Mn, Mg, Zn and (EtO)3SiH, had no reactivity (entry 18). Surprisingly, the reaction was sensitive to solvents: Amides, such as DMF and MeCONMe2, were viable media (entries 1 and 19), but other solvents, such as MeCN, 1,4-dixoane and ClCH2CH2Cl, were inert (entry 20). These results imply that amides may participate in the reaction besides as media. Decreasing temperatures led to diminishing yields (entry 21). The standard conditions were compatible with a scale up to 3 mmol 1a, giving 3aa in excellent yield (entry 22).
After confirming the optimized conditions, we set out to study the generality of this umpolung C—S radical reductive cross coupling protocol (Scheme 2). Gratifyingly, a variety of S-(trifluoromethyl)arylsulfonothioates 2b-i efficiently underwent the reaction with bromide 1a, Ni(ClO4)2·6H2O, L1 and Sn, affording 3ab-3ai in 85%–98% yields. Furthermore, several aryl functionalities, including 4-MeC6H4, 4-tBuC6H4, 4-MeOC6H4, 4-ClC6H4, 4-FC6H4, 4-CF3C6H4, naphthalen-2-yl and thiophen-2-yl, were well tolerated. Whereas using 2 h reacted with NiBr2 catalyst reduced the yield of 3ah to 53%.
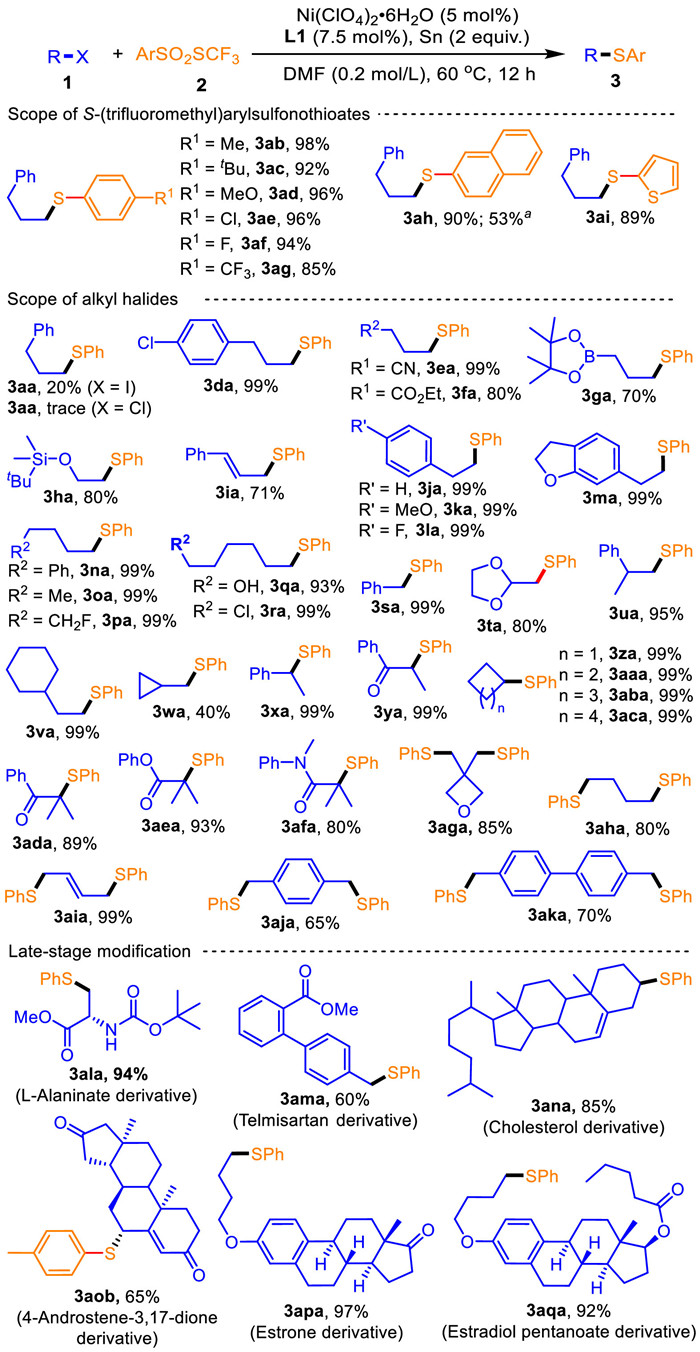
We next aimed to evaluate the scope of alkyl halides 1 (Scheme 2). Surprisingly, alkyl iodide, 3-phenylpropyl iodide 1b, was lower reactive for furnishing 3aa in 20% yield, attributing to readily decomposition of the C—I bonds. Using lower reactive 3-phenylpropyl chloride 1c failed to construct 3aa. Strikingly, a wide range of functionalized alkyl bromides 1d-aq accommodated to this umpolung C—S radical reductive cross coupling (3da-aqa). For example, functionalized propyl bromides 1d-g afforded 3da-ga, respectively, in 70%–99% yields where a functionality, such as 4-ClC6H4, CN, CO2Et, and 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl, at the position γ to the bromide atom was intact. This optimal conditions were compatible with (2-bromoethoxy)(tert-butyl)-dimethylsilane 1), producing the high useful silyl-substituted product 3 ha in 80% yield. Using (3-bromoprop-1-en-1-yl)benzene 1i, an alkene, furnished cinnamyl(phenyl)sulfane 3ia in good yield. The linear alkyl chains containing one to six carbon atoms were competent to the coupling, and several functional groups, including aryl, F, OH and Cl, were tolerated (3ja-sa). Alkyl bromides 1t-w with steric hindrance were suitable substates (3ta-wa). Broad secondary and tertiary alkyl bromides, including (1-bromoethyl)benzene 1x, α-bromoketones (1y, 1ad), four- to seven-membered cycloalkyl bromides (1z-ac), α-bromo ester (1ae) and α-bromo amide (1af), were subject to the coupling, furnishing the corresponding secondary and tertiary alkyl sulfanes 3xa-aca in high to quantitative yields. Interestingly, dual umpolung C—S radical reductive cross couplings of alkyl dibromides 1ag-ak executed successfully to access disulfanes 3aga-aka, which highlights the applicability of our protocol in organic and material synthesis. A number of natural product- or bioactive molecule-based alkyl bromides 1al-aq, such as L-alaninate derivative [69], telmisartan derivative [70], cholesterol derivative [71], 4-androstene-3,17-dione derivative [72], estrone derivative [73] and estradiol pentanoate derivative [74], exposed to the optimized conditions resulted in selective transformation of the C(sp3)—Br bonds to the C(sp3)—S bonds to produce highly valuable complex products 3ala-ana, 3aob, 3apa-aqa, thus providing a powerful route to selective late stage modification of complex bioactive substrates with multiple potential sites of reaction. Unfortunately, aryl halides, such as bromobenzene and iodobenezen, had no reactivity for the reaction.
In contrast to alkene-containing bromides 1i, 1ai, and 1an-ao (3ia, 3aia, 3ana, 3aob, Scheme 2), 6-bromohex-1-ene 1ar was converted to a mixture of the desired product 3ara and the intramolecular alkene difunctionalization product 4ara in 71% total yield with 5.1:1 chemoselectivity (Eq. 1, Scheme 3) [45]. Moreover, increasing concentrations of the Ni(ClO4)2·6H2O/L1 catalytic system shifted the chemoselectivity toward the coupling, and using 10 mol% Ni(ClO4)2·6H2O led to occurrence of the coupling exclusively. These radical clock experiments support that the reaction proceeds via a radical chain process [45,75-78]. Gratifyingly, 3-bromoprop-1-yne 3as was a suitable substrate, efficiently affording 3asd in 98% yield (Eq. 2). It was noted that no conversion of bromide 1a was observed in the absence of PhSO2SCF3 (Eq. 3), supporting initiation of this coupling not from the alkyl bromide component. Using PhSO2S(4-ClC6H4) 2j resulted in the selectivity toward direct C—S reductive cross coupling with the S(4-ClC6H4) moiety [45-49], not the PhSO2 moiety, to afford 3aj in 19% yield along with 4-chlorobenzenethiol 5j in 10% yield, but PhSO2SCH2(CH2)2Ph 2k had no reactivity (Eq. 4). The different chemoselectivity support that this current protocol performs a different mechanism from the previously reported reductive C—S cross coupling transformations [22,43-57], probably attributing to both the electron effect of the SCF3 group and reduction behavior of Sn. In the presence of Sn or Mn reductant, PhSSCF3 2l could react with bromide 1a to access 3aa in 10%–15% yields (Eq. 5), but leaving out each led to no detectable product 3aa. These results suggest that the PhSSCF3 is not the key intermediate during this current process, and the reductant can simultaneously assist both the S—S bond cleavage and the C—S bond formation. Subsequently, a series of the sulfur sources, including PhSSPh (2m), PhSO2CN (2n), PhSO2CF3 (2o), PhSO2F (2p), PhSO2Na (2q), PhSO3Na (2r), PhSO2N3 (2s) and PhSO2NCO (2t), were examined, but all had no reactivity under the optimized conditions (Eq. 6). It is noteworthy that both electrophilic PhSSPh (2m) and PhSO2CN (2n), the reported highly reactive thiolation reagents [43,44,48], are inert, thus ruling out the generation of PhSSPh as the key intermediate.
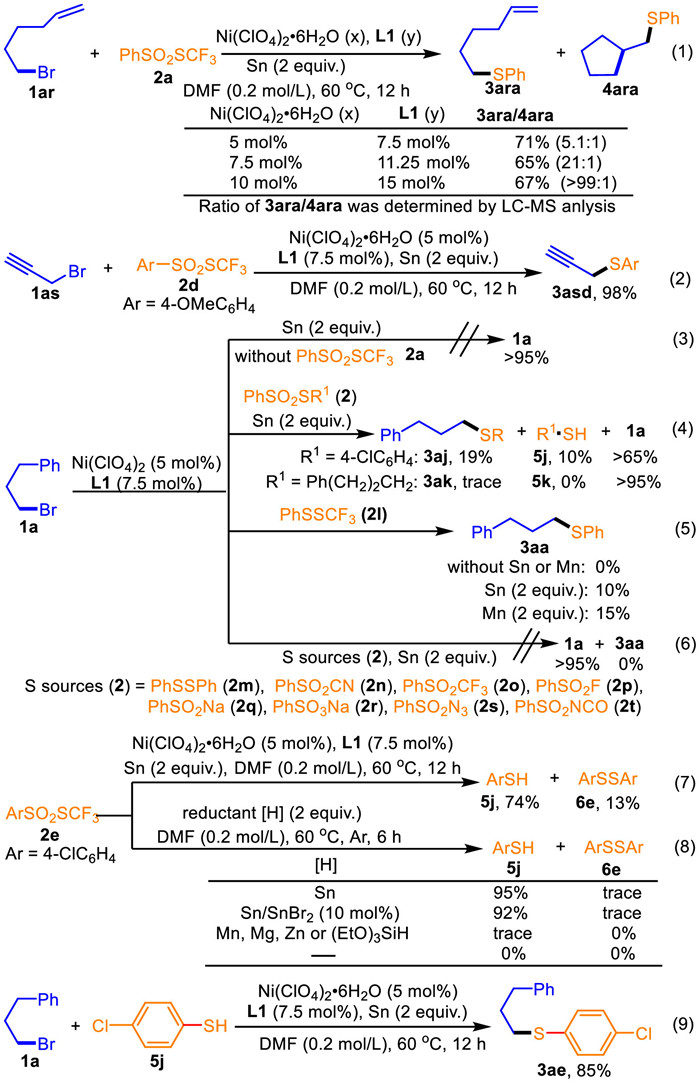
To further understand the mechanism, control reduction experiments with (4-ClC6H4)SO2SCF3 2e were conducted (Eqs. 7 and 8). In the presence of Ni(ClO4)2·6H2O, L1 and Sn, substrate 2e was reductively decomposed to 4-ClC6H4SH 5j in 74% yield and 4-ClC6H4SS(4-ClC6H4) 6e in 13% yield (Eq. 7). Reduction of (4-ClC6H4)SO2SCF3 2e with Sn run smoothly, affording 4-ClC6H4SH 5j exclusively in 95% yield; however, the SnBr2 additive (10 mol%) is detrimental and decreased the yield of 5j slightly to 92% yield (Eq. 8). The reason may be that the SnBr2 salt can promote the formation of disulfide [48], which would suppress the current coupling. It is noted that the reduction reaction is also sensitive to reductants: other reductants, such as Mn, Mg, Zn or (EtO)3SiH, are inert, and no reduction of the (4-ClC6H4)SO2SCF3 2e was observed without reductants (Eq. 8). These findings are consistent with the results observed in Table 1 (entries 1, 16 and 18), and support that the generation of the active benzenethiol-type intermediate, not the reported active PhSH and/or PhSSPh intermediates [22,43-57], is the key step. Under the optimized conditions, 4-ClC6H4SH 5j was less reactive than (4-ClC6H4)SO2SCF3 2e as using 4-ClC6H4SH 5j directly reacted with alkyl bromide 1a delivered a lower yield of 3ae (85% yield) (Eq. 9) than that of (4-ClC6H4)SO2SCF3 2e (96% yield, Scheme 2). It is because among the current coupling processes thermoinduced reduction of 4-ClC6H4)SO2SCF3 2e occurs to generate the higher reactive 4-ClC6H4S-based intermediate, not 4-ClC6H4SH 5j, to directly react with the active Ni species, thus avoiding further umpolung step of 4-ClC6H4SH to form the reactive 4-ClC6H4S-based intermediates (such as disulfides) and side reactions.
As shown in Scheme 4, the reaction of bromide 1a with PhSO2SCF3 2a was inhibited by radical scavengers, such as TEMPO, BHT and hydroquninone (Eq. 10). In the presence of TEMPO, the methylated products 8 and 8′ was detected by GC–MS analysis and no phenylpropyl-substituted product 7 from bromide 1a was observed (Eq. 9). Identical results were obtained from the reaction of 1a alone in the presence/absence of Ni(ClO4)2·6H2O and L1 (Eq. 11). These observations speculate that the methyl radical is generated from DMF, and DMF may really engage the umpolung C-S radical reductive cross coupling reaction. To verify thespeculations, control transformations of DMF with TEMPO were examined (Eq. 12). No reaction of DMF with TEMPO occurred when performing at 60 ℃ for 12 h. Using 2 equiv. Sn resulted in the formation of 8 in 3.7% GC yield. The optimized conditions that comprise a combination of 5 mol% Ni(ClO4)2·6H2O, 7.5 mol% L1 and 2 equiv. Sn were further confirmed, thus giving 8 in the highest 4.5% GC yield. Increasing loading of Ni(ClO4)2·6H2O and L1 led to diminishing GC yields of 8. The reason may be that the optimal loadings of the Ni(ClO4)2/L1 system efficiently initiate the generation of the radicals and effectively improve their reactivity, whereas the higher loadings of the Ni(ClO4)2/L1 system over activate the radicals to cause some unwanted side-reactions. The high reduction potentials of tin (Sn; −0.45 V vs. SCE) and DMF (−1.95 V vs. SCE) are proven to be useful reductants [79-83]. These findings indicate that thermoinduced reduction of DMF with Sn occurs to generate an amidyl radical anion intermediate [83-88], and DMF as an organic catalyst mediated the umpolung C-S radical reductive cross coupling reaction.

In the presence of 2 equiv. Sn, a stoichiometric amount of the Ni(Ⅱ) complex 9 (Eq. 13) exhibits identical catalytic activity to the NiBr2/L1 catalytic system (3ah, Scheme 2). However, neglecting Sn led to no detectable C—S cross coupling (Eq. 13). These observations prove the importance of the reduction process and the Ni0 species, not the NiⅡ salts, is the real active catalyst, which are further verified by the results using a stoichiometric amount of Ni(ClO4)2·6H2O (Eq. 14). The C—S radical reductive cross coupling of 1a with 2a and 1 equiv. Ni(ClO4)2·6H2O in the presence/absence of Sn (Eq. 14): Neglecting Sn caused no desired reaction after 12 h, but supplementing Sn to the same pot resulted in the formation of 3aa in 45% yield for 12 h.
Synthetic utilizations of phenyl(3-phenylpropyl)sulfane 3aa were conducted under oxidative conditions (Eq. 14) [22,43-57] Sulfane 3aa was converted to highly valuable ((3-phenylpropyl)sulfinyl)-benzene 10aa and ((3-phenylpropyl)-sulfonyl)benzene 11aa, respectively, in quantitative yields (Eq. 15). However, both substrates 10aa and 11aa could not be transformed to 3aa under the optimized conditions, excluding the possibility of the umpolung C—S radical reductive cross coupling via the 10aa and/or 11aa formation process.
Based on the current results and precedent literatures [22,43-57,75-88], the plausible mechanism for the Ni-catalyzed umpolung C—S radical reductive cross coupling reaction was proposed (Scheme 5). Initially, thermoinduced reduction of DMF with Sn affords an amidyl radical anion intermediate J [79-88]. Meanwhile, coordination of the NiⅡ species with the dinitrogen-based ligand L forms the active Ni0 species. Subsequently, the reaction of the active Ni0 species with the sulphydryl sulfur-centered radical (PhS·) intermediate F, which is formed from the umpolung reduction and single electron transfer (SET) of PhSO2SCF3 2a with Sn and the intermediate J, occurs to produce the LnNiⅠSPh intermediate G. Oxidation addition of the intermediate G with 3-phenylpropyl bromide 1a affords the Ph(CH2)2CH2(Ln)NiⅢBr(SPh) intermediate H, followed by reductive elimination of the intermediate H to give the LnNiⅠBr intermediate I and the desired product 3aa. Finally, reduction of the intermediate I by Sn regenerates the active Ni0 species to start a new catalytic cycle.
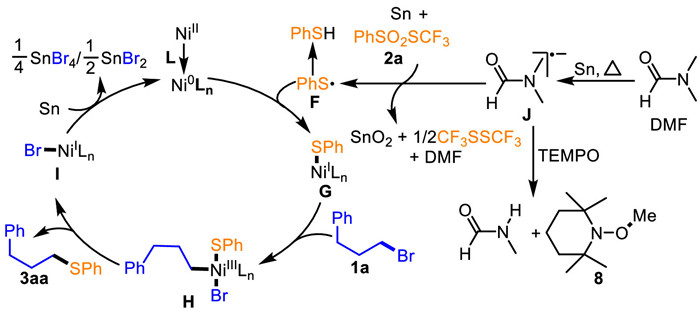
In summary, we have disclosed a novel catalytic radical reductive strategy for umpolung transformation of S-(trifluoromethyl)arylsulfonothioates via cooperative DMF and nickel reductive catalysis. This strategy was developed in a umpolung C—S radical reductive cross coupling of S-(trifluoromethyl)arylsulfonothioates with unactivated alkyl halides to assemble alkyl aryl thioethers. The reaction involves the formation of a sulfur-centered radical through thermoinduced umpolung reduction of S-(trifluoromethyl)arylsulfonothioates with DMF and Sn, as well as features excellent selectivity and wide functional group tolerance, which can be of great synthetic value for organic synthesis, such as applications in late-stage derivatization of pharmaceuticals and naturally occurring molecules, and creation of new reactions to access value-added derivatives of feedstocks. Mechanistic experiment evidence suggests that thermoinduced reduction of DMF by Sn readily occurs to generate the amidyl radical anion followed by umpolung reduction and SET of S-(trifluoromethyl)arylsulfonothioates with the amidyl radical anion and Sn to produce a sulfur-centered radical that engages a process of single electron oxidation of the active Ni0 species, unlike the previously explored alkyl carbon-centered radical counterparts.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the National Natural Science Foundation of China (No. 22271245), the Jiangxi Province Science and Technology Project (Nos. 20212AEI91002 and 20202ACBL213002) and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2021ZD01) for financial support.
A. Gangjee, Y.B. Zeng, T. Talreja, et al., J. Med. Chem. 50 (2007) 3046–3053. doi: 10.1021/jm070165j
H. Guo, B. Sun, H. Gao, et al., J. Nat. Prod. 72 (2009) 2115–2119. doi: 10.1021/np900654a
M. Feng, B. Tang, S.H. Liang, X. Jiang, Curr. Top. Med. Chem. 16 (2016) 1200–1216. doi: 10.2174/1568026615666150915111741
B. Banerjee, M. Koketsu, Coord. Chem. Rev. 339 (2017) 104–127. doi: 10.1016/j.ccr.2017.03.008
P. Devendar, G.F. Yang, Top. Curr. Chem. 375 (2017) 82. doi: 10.1007/s41061-017-0169-9
K.A. Scott, J.T. Njardarson, Top. Curr. Chem. 376 (2018) 5. doi: 10.1007/s41061-018-0184-5
N. Wang, P. Saidhareddy, X. Jiang, Nat. Prod. Rep. 37 (2020) 246–275. doi: 10.1039/c8np00093j
T. Kondo, T.A. Mitsudo, Chem. Rev. 100 (2000) 3205–3220. doi: 10.1021/cr9902749
I.P. Beletskaya, V.P. Ananikov, Chem. Rev. 111 (2011) 1596–1636. doi: 10.1021/cr100347k
C.F. Lee, Y.C. Liu, S.S. Badsara, Chem. Asian J. 9 (2014) 706–722. doi: 10.1002/asia.201301500
F. Dénès, M. Pichowicz, G. Povie, P. Renaud, Chem. Rev. 114 (2014) 2587–2693. doi: 10.1021/cr400441m
C. Shen, P. Zhang, Q. Sun, et al., Chem. Soc. Rev. 44 (2015) 291–314. doi: 10.1039/C4CS00239C
J. Li, S. Yang, W. Wu, H. Jiang, Org. Chem. Front. 7 (2020) 1395–1417. doi: 10.1039/d0qo00377h
N. Sundaravelu, S. Sangeetha, G. Sekar, Org. Biomol. Chem. 19 (2021) 1459–1482. doi: 10.1039/d0ob02320e
P. Annamalai, K.C. Liu, S.S. Badsara, C.F. Lee, Chem. Rec. 21 (2021) 3674–3688. doi: 10.1002/tcr.202100133
C. Zhu, H. Yue, J. Jia, M, Rueping Angew. Chem. Int. Ed. 60 (2021) 17810–17831. doi: 10.1002/anie.202013852
M. Kosugi, T. Shimizu, T. Migita, Chem. Lett. 7 (1978) 13–14. doi: 10.1246/cl.1978.13
C.G. Bates, R.K. Gujadhur, D. Venkataraman, Org. Lett. 4 (2002) 2803–2806. doi: 10.1021/ol0264105
F.Y. Kwong, S.L. Buchwald, Org. Lett. 4 (2002) 3517–3520. doi: 10.1021/ol0266673
K. Tanaka, K. Ajiki, Org. Lett. 7 (2005) 1537–1539. doi: 10.1021/ol0501673
M.A. Fernández-Rodríguez, Q. Shen, J.F. Hartwig, J. Am. Chem. Soc. 128 (2006) 2180–2181. doi: 10.1021/ja0580340
Y.C. Wong, T.T. Jayanth, C.H. Cheng, Org. Lett. 8 (2006) 5613–5616. doi: 10.1021/ol062344l
A. Correa, M. Carril, C. Bolm, Angew. Chem. Int. Ed. 47 (2008) 2880–2883. doi: 10.1002/anie.200705668
S.L. Buchwald, C. Bolm, Angew. Chem. Int. Ed. 48 (2009) 5586–5587. doi: 10.1002/anie.200902237
Y. Jiang, Y. Qin, S. Xie, et al., Org. Lett. 11 (2009) 5250–5253. doi: 10.1021/ol902186d
Y. Wang, L. Deng, X. Wang, et al., ACS Catal. 9 (2019) 1630–1634. doi: 10.1021/acscatal.8b04633
T.Y. Yu, H. Pang, Y. Cao, et al., Angew. Chem. Int. Ed. 60 (2021) 3708–3713. doi: 10.1002/anie.202013017
R.M. Oechsner, J.P. Wagner, I. Fleischer, ACS Catal. 12 (2022) 2233–2243. doi: 10.1021/acscatal.1c04895
J. Xia, R. Yao, M. Cai, Appl. Organometal. Chem. 29 (2015) 221–225. doi: 10.1002/aoc.3273
Y. Li, G. Bao, X.F. Wu, Chem. Sci. 11 (2020) 2187–2192. doi: 10.1039/c9sc05532k
R. Isshiki, M.B. Kurosawa, K. Muto, J. Yamaguchi, J. Am. Chem. Soc. 143 (2021) 10333–10340. doi: 10.1021/jacs.1c04215
Q. Tian, R. Sun, Y. Li, Org. Biomol. Chem. 20 (2022) 1186–1190. doi: 10.1039/d2ob00008c
K. Deng, A. Bensari-Bouguerra, J. Whetstone, T. Cohen, J. Org. Chem. 71 (2006) 2360–2372. doi: 10.1021/jo052535m
V.A. Vu, L. Bérillon, P. Knochel, Tetrahedron Lett. 42 (2001) 6847–6850. doi: 10.1016/S0040-4039(01)01426-5
A.H. Stoll, A. Krasovskiy, P. Knochel, Angew. Chem. Int. Ed. 45 (2006) 606–609. doi: 10.1002/anie.200501882
P.S. Herradura, K.A. Pendola, R.K. Guy, Org. Lett. 2 (2000) 2019–2022. doi: 10.1021/ol005832g
X. Xiao, M. Feng, X. Jiang, Angew. Chem. Int. Ed. 55 (2016) 14121–14125. doi: 10.1002/anie.201608011
P.S. Luo, M. Yu, R.Y. Tang, et al., Tetrahedron Lett. 50 (2009) 1066–1070. doi: 10.1016/j.tetlet.2008.12.066
I.M. Yonova, C.A. Osborne, N.S. Morrissette, E.R. Jarvo, J. Org. Chem. 79 (2014) 1947–1953. doi: 10.1021/jo402586v
Z.B. Dong, M. Balkenhohl, E. Tan, P. Knochel, Org. Lett. 20 (2018) 7581–7584. doi: 10.1021/acs.orglett.8b03319
S. Graßl, C. Hamze, T.J. Koller, P. Knochel, Chem. Eur. J. 25 (2019) 3752–3755. doi: 10.1002/chem.201806261
F. Zhu, Z. Chen, M.A. Walczak, J. Org. Chem. 85 (2020) 11942–11951. doi: 10.1021/acs.joc.0c01399
C. Zong, J. Liu, S. Chen, R. Zeng, J. Zou, Chin. J. Chem. 32 (2014) 212–218;. doi: 10.1002/cjoc.201300830
N. Li, J. Yao, L. Wang, et al., Inorg. Chem. Commun. 98 (2018) 99–104;. doi: 10.1016/j.inoche.2018.10.010
L. Wang, J. Qiao, J. Wei, et al., Tetrahedron 76 (2020) 130750;. doi: 10.1016/j.tet.2019.130750
Y. Fang, T. Rogge, L. Ackermann, et al., Nat. Commun. 9 (2018) 2240;. doi: 10.1038/s41467-018-04646-2
J. Li, W. Rao, S.Y. Wang, S.J. Ji, J. Org. Chem. 84 (2019) 11542–11552. doi: 10.1021/acs.joc.9b01387
J. Li, S.Y. Wang, S.J. Ji, J. Org. Chem. 84 (2019) 16147–16156. doi: 10.1021/acs.joc.9b02431
J.J. Ai, J. Li, S.J. Ji, S.Y. Wang, Chin. Chem. Lett. 32 (2021) 721–724. doi: 10.1016/j.cclet.2020.07.007
F. Wang, W. Rao, S.Y. Wang, J. Org. Chem. 86 (2021) 8970–8979. doi: 10.1021/acs.joc.1c00903
N.W.J. Ang, L. Ackermann, Chem. Eur. J. 27 (2021) 4883–4887. doi: 10.1002/chem.202005449
B. Gong, H. Zhu, Y. Liu, Green Synth. Catal. 3 (2022) 110–115. doi: 10.1016/j.gresc.2021.10.002
D. Yang, Q. Yan, E. Zhu, J. Lv, W.M. He, Chin. Chem. Lett. 33 (2022) 1798–1816. doi: 10.1016/j.cclet.2021.09.068
L. Qi, X. Pang, K. Yin, Chin. Chem. Lett. 33 (2022) 5061–5064. doi: 10.1016/j.cclet.2022.03.070
D. Liu, H.X. Ma, P. Fang, T.S. Mei, Angew. Chem. Int. Ed. 58 (2019) 5033–5037. doi: 10.1002/anie.201900956
M.T. Lan, W.Y. Wu, S.H. Huang, et al., RSC Adv. 1 (2011) 1751–1755. doi: 10.1039/c1ra00406a
T. Zhong, Z. Chen, J. Yi, G. Lu, J. Weng, Chin. Chem. Lett. 32 (2021) 2736–2750. doi: 10.1016/j.cclet.2021.03.035
H. Li, C. Shan, C.H. Tung, Z. Xu, Chem. Sci. 8 (2017) 2610–2615. doi: 10.1039/C6SC05093J
D. Zhu, X. Shao, X. Hong, et al., Angew. Chem. Int. Ed. 55 (2016) 15807–15811. doi: 10.1002/anie.201609468
Q. Zhao, L. Lu, Q. Shen, Angew. Chem. Int. Ed. 56 (2017) 11575–11578. doi: 10.1002/anie.201705633
H. Li, Z. Cheng, C.H. Tung, Z. Xu, ACS Catal. 8 (2018) 8237–8243. doi: 10.1021/acscatal.8b02194
S H., H. Li, T. Xie, et al., Org. Chem. Front. 6 (2019) 1663–1666. doi: 10.18410/jebmh/2019/336
K. Gadde, P. Mampuys, A. Guidetti, et al., ACS Catal. 10 (2020) 8765–8779. doi: 10.1021/acscatal.0c02159
M.Y. Wang, X.Q. Zhu, X.L. Zhang, et al., Org. Biomol. Chem. 18 (2020) 5918–5926. doi: 10.1039/d0ob01160f
J. Liu, H. Yao, X. Li, et al., Org. Chem. Front. 7 (2020) 1314–1320. doi: 10.1039/d0qo00343c
R. Pang, R. Yao, S. Lua, Y. Zhou, W. Chen, Chin. Chem. Lett. 32 (2021) 453–456. doi: 10.1016/j.cclet.2020.05.032
S. Chen, Q. Wen, Y. Zhu, Chin. Chem. Lett. 33 (2022) 5101–5105. doi: 10.1016/j.cclet.2022.04.022
X. Pannecoucke, T. Besset, Org. Biomol. Chem. 17 (2019) 1683–1693. doi: 10.1039/c8ob02995d
E. Del Carpio, L. Hernández, C. Ciangherotti, et al., Coord. Chem. Rev. 372 (2018) 117–140. doi: 10.1016/j.ccr.2018.06.002
M. Sharpe, B. Jarvis, K.L. Goa, Drugs 61 (2001) 1501–1529. doi: 10.2165/00003495-200161100-00009
H.C. Kwaan, A.J.S. Mcfadzean, Nature 179 (1957) 260. doi: 10.1038/179260a0
R.W. Brueggemeier, P.K. Li, Cancer Res. 48 (1988) 6808–6810.
A. Morsy, P.C. Trippier, J. Med. Chem. 62 (2019) 4252–4264;. doi: 10.1021/acs.jmedchem.8b01530
T. Niclo, R.S. Snell, Nature 179 (1957) 261.
J. Breitenfeld, J. Ruiz, M.D. Wodrich, X. Hu, J. Am. Chem. Soc. 135 (2013) 12004–12012. doi: 10.1021/ja4051923
S. Biswas, D.J. Weix, J. Am. Chem. Soc. 135 (2013) 16192–16197. doi: 10.1021/ja407589e
C. Zhao, X. Jia, X. Wand, H. Gong, J. Am. Chem. Soc. 136 (2014) 17645–17654. doi: 10.1021/ja510653n
C.E.I. Knappke, S. Grupe, D. Gärtner, et al., Chem. Eur. J. 20 (2014) 6828–6842. doi: 10.1002/chem.201402302
N. Pewnim, S. Roy, Electrochim. Acta 90 (2013) 498–506. doi: 10.1016/j.electacta.2012.12.053
A.P. Esteves, E.C. Ferreira, M.J. Medeiros, Tetrahedron 63 (2007) 3006–3009. doi: 10.1016/j.tet.2007.01.071
C.P. Andrieux, L. Gélis, M. Medebielle, J. Pinson, J.M. Savéant, J. Am. Chem. Soc. 112 (1990) 3509–3520. doi: 10.1021/ja00165a040
M.H. Baik, R.A. Friesner, J. Phys. Chem. A 106 (2002) 7407–7412. doi: 10.1021/jp025853n
N.L. Holy, J.D. Marcum, Angew. Chem. Int. Ed. 10 (1971) 115–124. doi: 10.1002/anie.197101151
J.M. Saveant, Acc. Chem. Res. 13 (1980) 323–329. doi: 10.1021/ar50153a005
A. Arora, J.D. Weaver, Acc. Chem. Res. 49 (2016) 2273–2283. doi: 10.1021/acs.accounts.6b00259
I.A. Shkrob, T.W. Marin, J. Phys. Chem. A 116 (2012) 1746–1757. doi: 10.1021/jp2115687
L. Sviatenko, L. Gorb, F. Hill, et al., Chem. Heterocycl. Compd. 50 (2014) 311–318. doi: 10.1007/s10593-014-1484-5
K.C. Mullane, T. Cheisson, E. Nakamaru-Ogiso, et al., Chem. Eur. J. 24 (2018) 826–837. doi: 10.1002/chem.201703396

Scheme 2 Variation of the alkyl halides (1) and arylsulfonothioate (2). Reaction conditions: 1 (0.2 mmol), 2 (0.22 mmol; 1.1 equiv.), Ni(ClO4)2·6H2O (5 mol%), L1 (7.5 mol%), Sn (2 equiv.), DMF (0.2 mol/L; 1 mL), argon, 60 ℃ and 12 h. a NiBr2 (5 mol%) instead of Ni(ClO4)2·6H2O.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:


