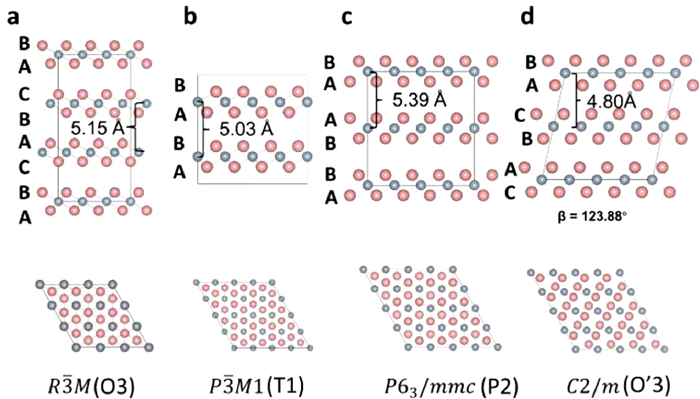
Figure 1.
The optimization structure of δ-MnO2. (a) R3M (O3 type). (b) P3M1 (T1 type). (c) P63/mmc (P2 type). (d) C2/m (O'3 type).

Facing the serious environmental pollution and massive consumption of fossil energy, the devices towards large scale energy storage is trying to use green energy for replacing the fossil energy sources [1-3]. Against the sporadic and unstable energy supply, rechargeable aluminum batteries (RABs), with theoretically high energy density and low cost, benefiting from multi-electron reaction is a hopeful device for energy storage [4,5]. In addition, due to the abundant resources of Al, it is easily to keep the prices of Al supplies stable, even in the case of large-scale utilization of Al minerals. Meanwhile, in order to maximize the advantages of RABs, cathode materials also tends to be cheaper and environment-friendly. Therefore, MnO2, an adequate metal oxide, become a candidate for matching the low cost anode.
MnO2 is reported to be a potential electrode material for different types of batteries such as Li-ion, Na-ion, Mg-ion, Zn-ion [6-9]. As for the battery system of Al-ion, Brooke Schumm Jr in 1976 and A. Sivashanmugam et al. in 2008 used aluminum-MnO2 dry cells of "D" size configuration for replacing the Lechlanche dry cell [10,11]. The two works were early humans using MnO2 as a cathode material in primary aluminum batteries. In recent years, MnO2 has gradually been used for rechargeable aluminum batteries, such as the experiments confirm the reversible insertion/extraction of Al-ion under the 1 mol/L Al(NO3)3 aqueous electrolyte, Mg-doped α-MnO2 cathode and "water-in-salt" aqueous AlCl3/MnSO4 electrolyte, α-MnO2 coated Mn2AlO4 cathode [12-16]. In addition, the experiment, an acid aqueous electrolyte of 1 mol/L Al(OTf)3, 1 mol/L Zn(OTf)3 and 0.1 mol/L MnSO4 or 1.3 mol/L Al(OTf)3, 0.3 mol/L Zn(OTf)3 and 0.3 mol/L MnCl2 demonstrates that MnO2 can be reversibly dissolved and deposited, allowing exploitation of the 2-electron, charge storage capacity of MnO2 with high cyclability and efficiency, during the role of [Al(H2O)6]3+ complex as a proton donor [17].
In fact, there are five phases of MnO2 polymorphs, which are α-MnO2, β-MnO2, γ-MnO2, δ-MnO2 and λ-MnO2 [18,19]. The connectivity of the fundamental octahedral unit MnO6, e.g., via the edges and/or corners, determines the structure of these MnO2 polymorphs. Layered compounds can store the carriers into the transition metal layers, providing a free room for ions diffusion. Hence, δ-MnO2 is a candidate for solve the sluggish dynamics of Al ions. According to the experiments, δ-MnO2 has two types of electrolytes for battery design, ionic liquids (ILs) and aqueous solutions. As for ILs, Paloma Almodóvar et al. [20] investigated δ-MnO2 (C2/m) nanofibers as cathode material and showed an initial discharge capacity of 59 mAh/g. And the cycle life is about 15 cycles at 37 mAh/g at a current rate of 100 mA/g. Martin Eckert et al. reports a high discharge voltage of 2.0–0.8 V in RABs [21]. Interesting, aqueous aluminum ion batteries (AAIBs) has delivered a remarkable energy density of 620 Wh/kg and keep a capacity of 320 mAh/g for 65 cycles [22]. However, since the electrolyte is an acidic electrolyte, the disproportionation reaction of Mn3+ and the reversible electrodissolution/electrodeposition makes δ-MnO2/Al cell more complex. The depositional δ-MnO2 phase is hard to representation, further increases the difficulty of mechanism investigated. However, the excellent electrochemical performance of AAIBs has been confirmed in experiments [17,22]. Therefore, the issues need to be solved to exploring the electrochemistry performance of δ-MnO2 in AAIBs and understanding the Al-ion diffusion mechanism of δ-MnO2. However, the current experimental methods have encountered obstacles to investigate. Instead, a first-principles calculation is successful to investigate the mechanism of ions insertion and intrinsic characteristics of materials, and these methods are supplements to experimental efforts for understanding the performance in molecular scale [23,24].
There are four space groups of δ-MnO2 (layered MnO2), which are R3M (O3 type), P3M1(T1 type), P63/mmc (P2 type) and C2/m (O'3 type) showed in Fig. 1 [25]. For the different space groups of MnO2, the stacking order of O layers is the key for different space groups of δ-MnO2. For O3 and O'3 type MnO2, the O layer is the order of "BACBAC…". T1 type of MnO2 is "BABA…". P2 type of MnO2 is "BAAB…". Although the difference between different types of δ-MnO2 lies in the location of the Mn and O ions, the small structure changes can lead to different results in Al3+ diffusion. Zhou et al. reported the 1T-MnO2 as potential surface of multivalent cation diffusion. Although the work indicated the excellent ion diffusion of Al, the drawback is that the JT (Jahn-Teller) distortion of Mn3+ are not considered and the only 1T-MnO2 has been investigated [26]. The published works have shown that JT distortion can influence the ion diffusion, even to the contrary conclusion [27]. Reviewing the current research, it is not clear for us to understand the difference of four different MnO2 types during the charging and discharging. Therefore, the molecular scale analysis is necessary to understand the difference after Al insertion.
For convenience, the discussion of δ-MnO2 describes by the O3 type, T1 type, P2 type and O'3 type, respectively, and the space groups are marked in diagrams. In this work, we investigated the four types of δ-MnO2 and understand the intrinsic electrochemical characteristics from atoms structure, electronic structure and ions diffusion. The voltage of Al insertion into δ-MnO2 is T type > O type > P type. However, the energy barriers of Al in T type and O type are 1.00 and 1.08 eV indicated a sluggish diffusion. Interesting, the energy barrier of Al migration is only 0.56 eV, due to a long (spacious pathways) and stable (2.007–2.030 Å) Al-O bonds in P2 type. By eliminated the influence of Mn3+, means the Mn3+ is far away with the diffusion ion, the energy barrier of Al migration could reach 0.19 eV in P2 type. It confirms the obviously effect of Mn3+ polaron. Hence, key influencing factors of slow Al-ions diffusion are narrow pathways, unstable Al-O bonds and Mn3+ type polaron have been identified. Besides, due to the aqueous electrolyte, the water cannot avoid in the experiment, resulting in a large interlaminar spacing of ~7 Å [21]. However, the dehydration of δ-MnO2 makes the interlaminar spacing decrease to 5.5–5.6 Å [28,29]. The interlayer spacing determines the room of the carrier diffusion. Therefore, we also investigate the influence of large interlayer spacing (6.5 Å [29]) during the Al insertion/extraction. It is discovered that large interlayer spacing is beneficial to ions diffusion. These discover provide more evidence for researchers in developing a high energy density cathode material.
The Vienna ab initio simulation package (VASP) based on density functional theory (DFT) with the Projector Aleugmented Waves (PAW) method was used throughout our calculations [30,31]. A spin-polarized generalized gradient approximation (GGA) and the plane wave basis projector augmented wave (PAW) methods are used to solve the Kohn-sham equations with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional [31-33]. The valence configurations of Mn (3d5, 4s2), O (2s2, 2p4), Mg (3s2), Na (3s1) and Al (3s2, 3p1) orbital are considered in our calculations. Because of strong on-site electron-electron correlations among the Mn-3d electrons, the GGA+U method has been employed and the Ueff value is set as 3.9 eV [34-37]. The Monkhorst-Pack scheme with 3 × 3 × 2 k-point mesh is used for supercell [38]. The model of O3 (3 × 3 × 1 supercell), T1 (3 × 3 × 3 supercell) and O'3 type of MnO2 (3 × 3 × 1 supercell) contains a supercell with 54 O atoms and 27 Mn atoms. The model of P2 contains a 3 × 4 × 1 supercell with 48 O atoms and 24 Mn atoms. The optimized lattice parameters of δ-MnO2 in four different space groups are showed in Table 1. The cutoff energy for the expansion of plane wave function is 500 eV. During the optimization, all the atomic positions are fully relaxed and the final force between all the atoms is less than 0.01 eV/Å. And the energy converged to 10−7 eV/Å. To describe the effect of van der Waals interaction, the DFT-D3 method was considered in the ions adsorption, not in Al insertion into δ-MnO2 [39]. Besides, the Al insertion into a 14 Å vacuum layer was used for supercell to avoid unwished interaction between the two MnO2 layers on monolayer MnO2. To investigate the diffusion pathways of Al ions in MnO2, the NEB approach is employed by using 5 images from the starting to the ending most stable position [40,41].
In addition, the adsorption energy of metal ions is calculated by the follow formula:
|
|
(1) |
where Ead is the adsorption energy of metal ion on the monolayer MnO2. Emetal−MnO2, Emetal and EMnO2 are the energy of metal ion on the monolayer MnO2, the binding energy of bulk metal and the energy of monolayer MnO2, respectively.
The δ-MnO2 exhibits four structure classification, distinguishing by the stacking order of Mn-O layers. According to previous reports, it is unfavorable for Al ion adsorption on graphene [42]. In order to understand the adsorption of Al ions and multi-electron reaction of Al on the MnO2 surface, the Al ions' adsorption on the monolayer MnO2 is estimated. Similar to common layered materials, such as graphene, MoS2, phosphorene, the weak van der Waals force cannot securely bind the MnO layers, leading to the monolayer MnO2 from bulk MnO2 [43-45]. In 2003, the monolayer MnO2 had been synthesized by Yoshitomo Omomo et al. in experiment [46]. In addition, the monolayer MnO2 also reflects the electrochemical properties of MnO2 (001) surface, which is facing to the electrolyte. In order to better understand the multi-electron reaction of Al, the Na+ and Mg2+ are added as comparisons and the same-period elements could remit the effect of different ionic radius on performance.
According to previous works and our tests in Fig. S2 (Supporting information), the two types of adsorption sites are choose in the calculations, which are located above O and transition metal, named O-hollow site and M-hollow site in this paper, respectively [47]. All the ions are fullly optimized without fixation in the calculations. And the structures of Al ions' adsorption are shown in Fig. 2. According to the crystal field theory, the 3d orbital of Mn3+ splits into two parts, t2g orbital and eg orbital. The last electron of Mn3+ occupies the higher eg orbital forming the Jahn-Teller distortion. Due to the low Gibbs free energy of JT effect in Table S1 (Supporting information), all structures are taken into account the JT effect of Mn3+, except P2-type MnO2. On the M-hollow position, the distance between O and Na is 1.757 Å, which are largest among the distance of Al and O (1.248 Å), the distance of Mg and O (1.306 Å). Besides, on the O-hollow position, the distance of Na and O, Mg and O, Al and O are 1.634, 1.121 and 1.064 Å, respectively. Compared the distance of O-hollow position with the distance of M-hollow position, the distance between metal ions and O in the O-hollow position is smaller. Hence, the adsorption energy in the O-hollow position is larger than the M-hollow position in Table 2, indicating the strong adsorption force. Therefore, the stable adsorption sites of metals are O-hollow positions. In particular, as the chemical valence of metal ions increased, the high electron transfer, the distance between metal ions and O decreased, the adsorption energy of ions become smaller in harmonization of electromagnetic forces.

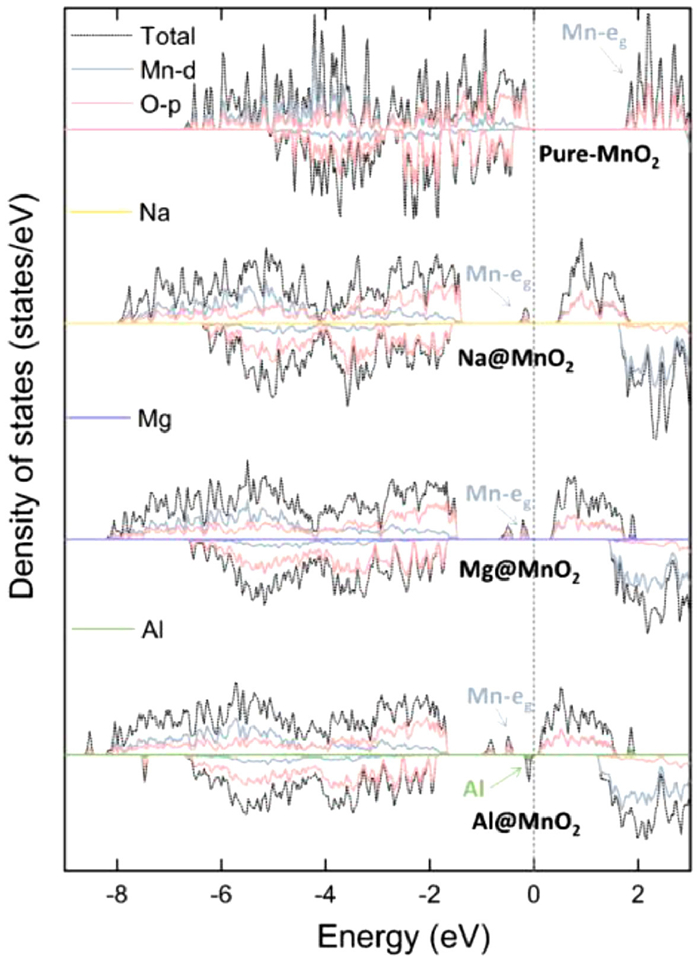
Due to the special electron configuration (3s23p1) of Al, when one electron loses from 3p1 orbital, Al has a stable electron configuration (3s23p0). In the previous work of our group, it is proved that the special electron configuration of Al makes the charge transfer between Al and borophene achieve one electron [48]. The electrons in 3s orbital is stable. Hence, it's not a multi-electron reaction. In addition, excellent electronic conductivity is also an important performance of electrode materials. The electronic conductivity of MnO2 has shown by examining the electronic structure of monolayer MnO2. In Fig. 3, the pure monolayer MnO2 has a band gap of 1.71 eV. After the Na, Mg and Al ions are adsorbed on the monolayer MnO2, the electron transfer from metal ions to MnO2, Mn-eg orbital gained valence electrons from the metal ions. Hence, the part of the Mn-eg orbital is below the Fermi energy level, leading to the small band gaps. The band gaps of ions adsorption on the MnO2 are 0.41, 0.29 and 0.37 eV for Na, Mg and Al adsorption, respectively. In addition, the 3s orbitals of Mg and Na are empty in Fig. 3, means the valence electrons of ions completely transfer from 3s orbitals to monolayer MnO2. Interesting, the part of Al-s and Al-p orbital is below the Fermi energy level, when Al adsorbed on the MnO2, indicating that the valence electrons of Al are not completely lost. Comparing the DOS of Mg@MnO2 with Al@MnO2 in Fig. 3, the electronic structures of Mn are same, two peaks of Mn-eg below the Fermi level. And the Bader calculation proves the 1.78|e| of Al and of 1.68|e| of Mg have been transferred. Hence, there are two Mn ions on the MnO2 forming two Mn3+, proved by the magnetic moment of Mn. In a word, although the adsorption energy of Al ions is high enough to store Al ions, the electron transfer of 3|e| cannot be achieved, limiting the capacities of RABs.
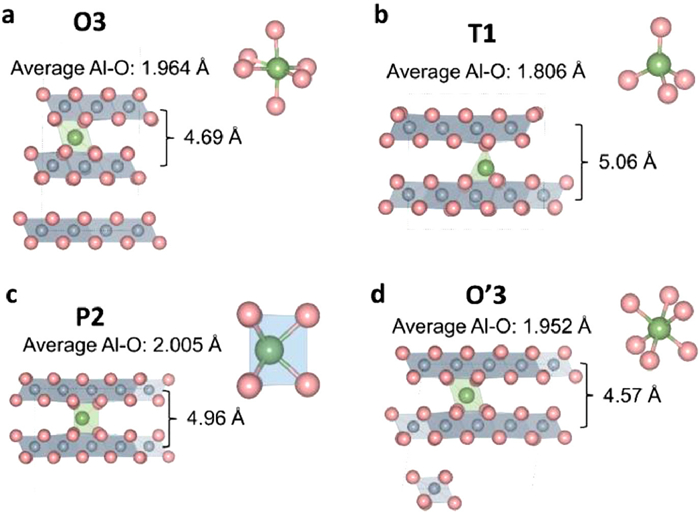
In order to break though the electron transfer of Al on monolayer MnO2, we investigated Al insertion into bulk MnO2 to achieve the 3|e| electron reaction of Al. In Fig. 1, the δ-MnO2 has four types, named O3, T1, P2 and O'3, respectively. The distance of Mn-Mn layers are 5.15, 5.03, 5.39 and 4.80 Å for O3, T1, P2 and O'3 types, respectively. In our works, after relaxing all the atoms, the β angle of O'3 type is 123.88° After comparing the Gibbs free energy of Al insertion with different structures, the best positions of Al insertion are showed in Fig. 4 because of the lowest structure energy. The details are showed in Table S2 (Supporting information). The optimal Al insertion position in O3 and O'3 types are the octahedral positions, the best position in P2 type is the center of triangular prism. Interesting, the best Al insertion position in T1 type is the tetrahedral position. In addition, for the O3, T1, P2 and O'3 types, the average bonds of Al-O are 1.964, 1.806, 2.005 and 1.952 Å, respectively. When Al inserted into layered MnO2, the interlayer spacing of Mn-Mn layers decreased to 4.69, 5.01 and 4.57 Å for O3, P2 and O'3, respectively, except T1 type, indicated the stronger interaction between Al and O.
According to previous reports, in experiments, the two discharge plateau of δ-MnO2 are Mn4+ to Mn3+ and Mn3+ to Mn2+ when Al insertion into MnO2 [15,20,26,49]. And the main discharge plateau of Mn4+ to Mn3+ is 87% of experimental capacities (279.17 mAh/g of 320 mAh/g [28]). Besides, due to the disproportionation reaction of Mn3+ and the dissolution of Mn2+, MnO2 causes phase transitions and collapses of structures [50,51]. However, the physical coating and ions doped avoid the reactions between the acid electrolyte and MnO2, alleviating phase transitions and structure collapse [50,52]. In order to accurately understand the electronic structure and voltage of Al insertion into layered MnO2, avoiding the effect of Mn2+, the main discharge plateau of Mn4+ to Mn3+ is discussed in this work to estimate the main voltage value and electronic structure of MnO2.
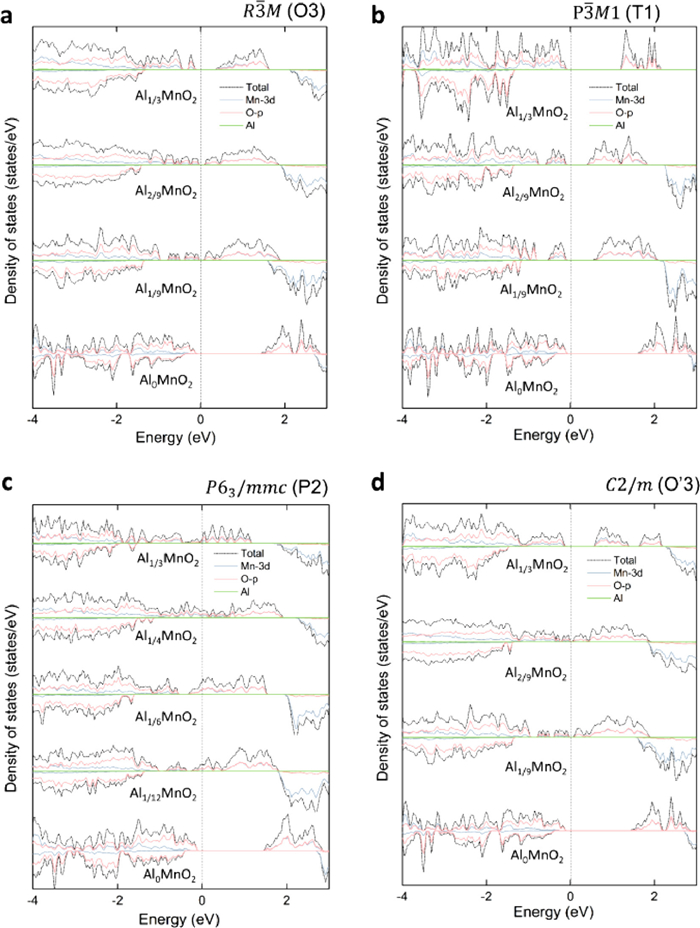
In Fig. 5, before the Al ion inserted into MnO2, layered MnO2 had typical semiconductor characteristics, and the band gap of 1.71 eV made the electron conductivity of MnO2 have to be assisted by some conductive agents. When Al inserted into MnO2 (Al0.111MnO2 for O3, T1 and O'3, Al0.083MnO2 for P2) initially, the P2 type presents an obvious metallic state with a good electronic conductivity, while the T1 type has obvious semiconducting characteristics. Besides, the band gap of T1 is obviously reduced. Particularly, due to the split of Mn-eg, the band gaps of O3, T1 and O'3 are narrow than the initial MnO2. With the further increase of the Al concentration to Al0.333MnO2, the Mn-eg orbitals in the O3 and O'3 types constantly gain electrons from Al, and the energy gap increased. At last, the Mn4+ completely transformed into Mn3+, the band gaps of Al0.333MnO2 become 0.29, 1.16 and 0.48 eV for O3, T1 and O'3, respectively. However, the band gaps of Al0.333MnO2 is smaller than pure MnO2. Interesting, the MnO2 of the P2 type always has metallic state regardless of the Al concentration. Besides, in the T1 type, the band gap decreases to 0.32 eV of Al0.222MnO2, keeping on the typical semiconductor characteristics. Until the Al concentration reaches Al0.333MnO2, the band gap becomes 1.13 eV. The energy gap is derived from the energy level difference between dz2 and dx2−y2. In Fig. 5, there is no electronic state of Al around the Fermi level, indicated the Al is complete oxidation. Further analyzing the magnetic moment of Mn, it shows that all the valence electron of Al transfer to Mn. Hence, the electron transfer could reach 3|e|, and the Al ions are ionic states.
The open circuit voltage is an important parameter for estimating the performance of batteries. Improving the voltage of RABs can increase the energy density of batteries. In our work, we calculate the voltage of Al insertion into MnO2 by using the following formula:
|
|
(2) |
where Einsertion-xAl is the energy of x Al insertion into MnO2, Einsertion-yAl is the energy of y Al insertion into MnO2, EAl is the energy of Al metal, x and y is the number of Al ions. The average voltages are shown in Table 3. Hence, the voltage of the T1 type is the highest of 2.56 V. The voltage of the P2 type is only 1.82 V. And the voltages of the O3 and O'3 types are 2.28 V and 2.18 V, which is fit with the experiment (O'3 type of 2.1 V) [20]. In addition, during the discharge of Mn4+ to Mn3+, the average voltages are 2.08 V, 2.11 V, 1.76 V and 2.06 V for O3, T1, P2 and O'3 type, respectively. It was found that the O type MnO2 has the high voltage, over 2 V, while the voltage of P type MnO2 is less than 2 V. And the average voltages are T type > O type > P type.
Due to the high valence of Al, the strong force between Al and O leads to a sluggish diffusion of ions in cathode materials, which hampers the application of RABs. Although, Zheng et al. proposed measures, such as widening diffusion channel and choosing flat potential energy surface, to promote the diffusion of Al after summarizing the published research works [53]. It is undeniable that the diffusion of Al ions is still a major obstacle to the development of RABs. Therefore, investigating the diffusion of Al ions is an unavoidable issue for improve the performance of RABs. After evaluated the diffusion of Al ions in the four types by DFT calculations, Figs. 6a and b showed the energy barriers of Al ions in O3 and T1 type are 1.08 eV and 1.00 eV, respectively. When Al inserted into the P2 types, the diffusion barrier of Al ions is only 0.56 eV in Fig. 6c. Facing the high charge characteristics of Al3+, the diffusion of ions is faster than Na ions in the layered material, Na2MnO3, indicating that P2 type is beneficial for ions diffusion [54]. However, Fig. 6d showed that the energy barrier of Al ions in the O'3 type was the highest, reaching 2.41 eV, indicated the difficult diffusion of Al ions in the intercalation.

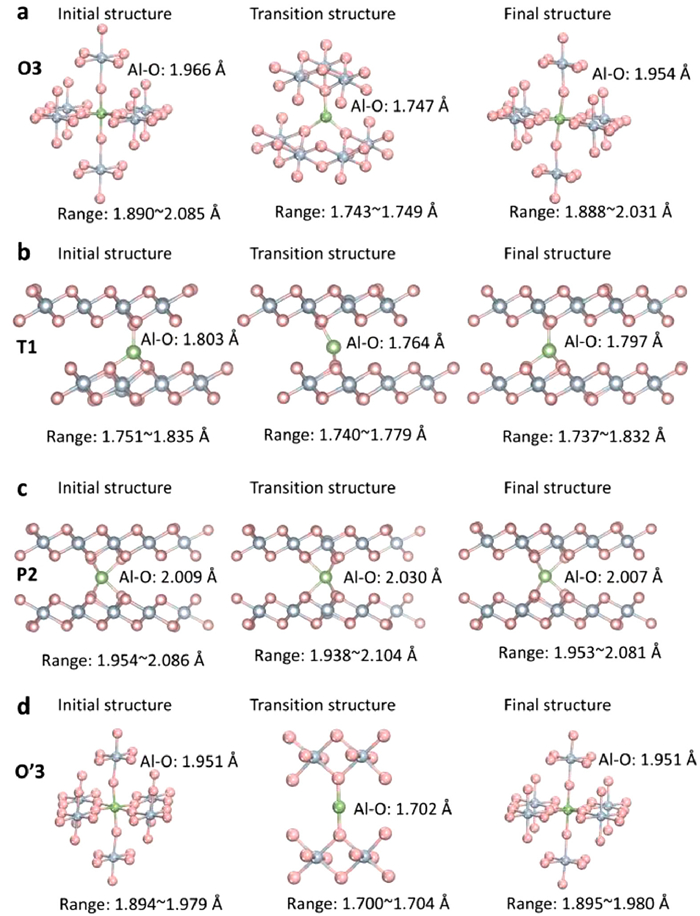
To understand the mechanisms of ions diffusion in δ-MnO2, observing the transition structure of ion diffusion in Fig. 7, the diffusion of Al ions in T1 is from tetrahedron sites to trigonal site to tetrahedron sites. However, the diffusion of Al ions in O3 is from octahedral sites to triangle center site to octahedral sites. Further observed the structure of the transition state in Fig. 4d, it should be noticed that the interlayer spacing of Mn-Mn layers is only 4.57 Å. The narrow interlayer spacing makes the length of Al-O bonds 1.702 Å be shorter than 1.951 Å of ground states before diffusion in Fig. 7d. Hence, at the effect of narrow interlayer spacing and short bonds, the strong force between Al and O increases the difficulty of Al ion diffusion. In addition, the Al-O bonds of transition structure in O3 and T1 are 1.747 Å and 1.764 Å. As for the ions diffusion of O3 and T1 types, considering the larger interlayer spacing of Mn-Mn slabs, the energy barriers of Al ions in O3 (1.08 eV) and T1 (1.00 eV) are less than O'3 type (2.41 eV). Interestingly, in P2 type, the transition structure is showed in Fig. 7c. The Al ions are always located in the center of the triangular prism. And the average bonds of Al-O are 2.009, 2.030 and 2.007 Å from initial structure to transition structure to final structure, respectively. The stable and long Al-O bonds showed a fast Al diffusion due to the stable potential energy surface. To understand the influence of Mn3+ during the Al migration, the Mn3+ has been eliminated by getting rid of 3|e| in the calculation. And the energy barrier of Al migration could reach 0.19 eV in P2 type in Fig. 8a. Therefore, it confirms the effect of Mn3+ type small polaron is obvious. To improve the ions diffusion, restraining the Mn3+ type small polaron need to be studied.

However, in most cases of experiments, the layered MnO2 exists H2O in the aqueous AIBs. Hence, the influence of water during the Al ions' diffusion cannot be ignored. Because the actual structure of H2O is complex and numerous. It is difficult to show the practical effect of H2O insertion into δ-MnO2. In next calculation, our group explores the effect of the large interlayer spacing causing by H2O in the Al ions' diffusion. In Na-ion batteries, the co-intercalation of H2O and Na brings about a large interlayer spacing, which makes the interlayer spacing of the Mn-Mn layer reach 5.5–5.6 Å, or even more than 7 Å [22,29,49]. In a published paper, Lucht et al. calculated the co-intercalation of H2O and Al in MnO2 and showed the interlayer spacing of Mn-Mn layers could reach 6.5 Å [29]. Hence, the fixed interlayer spacing of 6.5 Å is employed in the calculations (Fig. 8). The DOS is showed in Fig. 8b and Al ion adsorbed on the top layer of Mn-Mn layers in Fig. 8c. The DOS shows the Al@MnO2 has a good electron conductive and the Al ions is ionic state. In order to show the influence among the Mn-Mn layer during the diffusion of Al ions, we added the diffusion of Al ions on the monolayer as a control group. In Fig. 8d, it is showed that the energy barrier of Al ions' diffusion reached 0.84 eV in the large interlayer spacing of Mn-Mn layers, which has excellent ions diffusion. In addition, the diffusion barrier of Al ions in the monolayer reaches 1.78 eV, which is much higher than that of Al and H2O insertion into the MnO2 layer. Further observe the transition structure of Al diffusion in 6.5 Å interlayer spacing in Fig. 8c, when Al ions migrate, due to the attraction of Al and O (bottom layer), the distance of Al and O (bottom layer) decrease from 3.650 Å to 2.910 Å. Hence, the average Al-O bonds increase from 1.761 Å (three bonds) to 1.782 Å (two bonds) weakened the force between Al and O. Consequently, the MnO2 layer balance the force between Al and O (Top layer), promoting the Al ions diffusion. Comparing the O3 type and 6.5 Å interlayer spacing, the energy barrier decreases to 0.84 eV. Paloma Almodovar et al. synthesized O'3 MnO2. In the aluminum ion battery, the interlayer spacing of MnO2 reached ~7 Å, which was fit with the calculation simulation [20,22]. And MnO2 achieves intercalation and deintercalation of the aluminum species at large layer spacing, achieving a reversible capacity of 37 mAh/g [20], verified the effective strategy of increased the interlayer spacing. Therefore, the insertion of H2O can effectively reduce the diffusion barriers of Al ions and increase the diffusion speed of Al ions. Of course, this case is a simplified representative of Al ions insertion into crystal water of MnO2. However, it reflects the effect of larger interlayer spacing during the Al ions' diffusion.
In summary, this work provides a systematic and comprehensive assessment of four type layered MnO2 (O3, T1, P2 and O'3 type) as cathode materials in RABs, and by atomic structures, electronic structures, and diffusion of Al ions analysis, the suitable P2-type layered MnO2 has been verified to apply for RABs. As for O3 and T1 type MnO2, the higher open-circuit voltage promotes the MnO2 to achieve the higher energy density at the expense of charging/discharging speed. Regarding the O'3 type layered MnO2, the higher energy barrier of 2.41 eV indicates extremely slow ions diffusion and the key issues for application is to improve the dynamics. Base on the previous work for 2D materials, by comparing the carriers adsorption (Na, Mg, Al) of different chemical valence, the multi-electron reaction of Al in monolayer MnO2 has been investigated and the results shows the electron transfer has not reached 3|e|, means incomplete multielectron reaction. Given the key issue proposed, multilayer MnO2 could achieve a 3|e| multielectron reaction, which reduce the cost of production and advise researchers keep more multilayer MnO2 structure. To solve the issue of slow diffusion, further analysis of the mechanism of Al ion diffusion, the main reason of Al slow diffusion in O'3 is unstable and short Al-O bonds. Besides, the distance of Mn-Mn layers decrease because of Al insertion, reduced the room of Al migration. Comparing the T1 and O3 type MnO2, O3 type MnO2 has more spacious pathways and T1 type MnO2 has more stable Al-O bonds. Hence, the energy barriers of Al diffusion are similar. Interesting, the P2-type MnO2 keep a low energy barrier of 0.56 eV, due to a long and stable Al-O bonds of 2.007–2.030 Å Specially, the experiment cannot avoid water into layered MnO2. After considering the huge interlayer spacing of Mn-Mn layers causing by water, increasing the interlayer spacing can accelerate the diffusion of Al ions, promoting the application of O'3 types MnO2. In addition, if the Mn3+ is far away from the diffusion ions, the energy barrier of Al diffusion is reduced to 0.19 eV. It confirms the obviously effect of Mn3+ polaron. Therefore, there are three main reasons for slow ions diffusion that are narrow pathways, unstable Al-O bonds and Mn3+ type polaron. Although the average voltage of P2 (1.76 V) is the lowest than others, considering comprehensive performance, our works suggest that layered MnO2 should keep more P2-type MnO2 in the synthesis of materials for RABs, further developing the cathode materials of RABs and paving the way for large-scale energy storage.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported financially by the National Natural Science Foundation of China (No. 22075028).
J.B. Goodenough, Energy Storage Mater. 1 (2015) 158–161. doi: 10.1016/j.ensm.2015.07.001
J.B. Goodenough, Nat. Electron. 1 (2018) 204 204. doi: 10.1038/s41928-018-0048-6
K. Mizushima, P.C. Jones, P.J. Wiseman, et al., Mater. Res. Bull. 15 (1980) 783–789. doi: 10.1016/0025-5408(80)90012-4
F. Wu, H. Yang, Y. Bai, et al., Adv. Mater. 31 (2019) 1806510. doi: 10.1002/adma.201806510
Z. Huang, W. Wang, W.L. Song, et al., Angew. Chem. Int. Ed. 61 (2022) e202202696. doi: 10.1002/anie.202202696
F. Wu, X. Gao, X. Xu, et al., ChemSusChem 13 (2020) 1537–1545. doi: 10.1002/cssc.201903006
C. Yang, M. Han, H. Yan, et al., J. Power Sources 452 (2020) 227826. doi: 10.1016/j.jpowsour.2020.227826
Y. Wang, J. Liu, B. Lee, et al., Nat. Commun. 6 (2015) 6401. doi: 10.1038/ncomms7401
Z. Liu, X. Li, J. He, et al., J. Energy Chem. 68 (2022) 572–579. doi: 10.1016/j.jechem.2021.12.016
A. Sivashanmugam, S.R. Prasad, R. Thirunakaran, et al., J. Electrochem. Soc. 155 (2008) A725–A728. doi: 10.1149/1.2960897
B. Schumm, J. Electrochem. Soc. 123 (2019) 1696–1698.
J. Joseph, J.F.S. Fernando, M.A. Sayeed, et al., ChemElectroChem 8 (2020) 1048–1054.
W. Pan, J. Mao, Y. Wang, et al., Small Methods 5 (2021) 2100491. doi: 10.1002/smtd.202100491
A. Ejigu, L.W. Le Fevre, A. Elgendy, et al., ACS Appl. Mater. Interfaces 14 (2022) 25232–25245. doi: 10.1021/acsami.1c23278
Y. Wang, K.L. Ng, T. Dong, et al., Electrochim. Acta 405 (2022) 139808. doi: 10.1016/j.electacta.2021.139808
X. Zhang, W.L. Song, M. Wang, et al., Energy Storage Mater. 45 (2022) 586–594. doi: 10.1016/j.ensm.2021.12.020
V. Balland, M. Mateos, A. Singh, et al., Small 17 (2021) 2101515. doi: 10.1002/smll.202101515
M.F. Dupont, A.D. Cross, A. Morel, et al., J. Electrochem. Soc. 160 (2013) A1219–A1231. doi: 10.1149/2.012308jes
T. Hatakeyama, N.L. Okamoto, T. Ichitsubo, J. Solid State Chem. 305 (2022) 122683. doi: 10.1016/j.jssc.2021.122683
P. Almodóvar, D.A. Giraldo, J. Chancón, et al., ChemElectroChem 7 (2020) 2102–2106. doi: 10.1002/celc.202000425
M. Eckert, W. Peters, J.F. Drillet, Materials 11 (2018) 2399. doi: 10.3390/ma11122399
S. He, J. Wang, X. Zhang, et al., Adv. Funct. Mater. 29 (2019) 1905228. doi: 10.1002/adfm.201905228
H.R. Yao, P.F. Wang, Y. Gong, et al., J. Am. Chem. Soc. 139 (2017) 8440–8443. doi: 10.1021/jacs.7b05176
S.Q. Shi, J. Gao, Y. Liu, et al., Chin. Phys. B 25 (2016) 018212. doi: 10.1088/1674-1056/25/1/018212
C. Delmas, C. Fouassier, P. Hagenmuller, Phys. B+C 99 (1980) 81–85. doi: 10.1016/0378-4363(80)90214-4
B. Zhou, Z. Zhou, B. Li, et al., J. Phys. D: Appl. Phys. 54 (2021) 115303. doi: 10.1088/1361-6463/abcf75
L. Zheng, Z. Wang, M. Wu, et al., J. Mater. Chem. A 7 (2019) 6053–6061. doi: 10.1039/C8TA11955D
K.P. Lucht, J.L. Mendoza-Cortes, J. Phys. Chem. C 119 (2015) 22838–22846. doi: 10.1021/acs.jpcc.5b07860
X. Zhao, L. Mao, Q. Cheng, et al., Energy Storage Mater. 38 (2021) 397–437. doi: 10.1016/j.ensm.2021.03.005
G. Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 11169–11186. doi: 10.1103/PhysRevB.54.11169
P.E. Blochl, Phys. Rev. B 50 (1994) 17953–17979. doi: 10.1103/PhysRevB.50.17953
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758–1775.
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Y.F. Li, S.C. Zhu, Z.P. Liu, J. Am. Chem. Soc. 138 (2016) 5371–5379. doi: 10.1021/jacs.6b01768
V.V. Anisimov, J. Zaanen, O.K. Andersen, Phys. Rev. B 44 (1991) 943–954. doi: 10.1103/PhysRevB.44.943
F. Zhou, M. Cococcioni, C.A. Marianetti, et al., Phys. Rev. B 70 (2004) 235121. doi: 10.1103/PhysRevB.70.235121
W. Zuo, J. Qiu, X. Liu, et al., Nat. Commun. 11 (2020) 3544. doi: 10.1038/s41467-020-17290-6
J.D. Pack, H.J. Monkhorst, Phys. Rev. B 16 (1977) 1748–1749. doi: 10.1103/PhysRevB.16.1748
S. Grimme, J. Antony, S. Ehrlich, et al., J. Chem. Phys. 132 (2010) 154104. doi: 10.1063/1.3382344
G. Henkelman, H. Jónsson, J. Chem. Phys. 113 (2000) 9978–9985. doi: 10.1063/1.1323224
G. Henkelman, B.P. Uberuaga, H. Jónsson, J. Chem. Phys. 113 (2000) 9901–9904. doi: 10.1063/1.1329672
M.L. A. giorgousis, Y.Y. Sun, S. Zhang, ACS Energy Lett. 2 (2017) 689–693. doi: 10.1021/acsenergylett.7b00110
A.K. Geim, K.S. Novoselov, Nat. Mater. 6 (2007) 183–191. doi: 10.1038/nmat1849
B. Radisavljevic, A. Radenovic, J. Brivio, et al., Nat. Nanotechnol. 6 (2011) 147–150. doi: 10.1038/nnano.2010.279
A. Carvalho, M. Wang, X. Zhu, et al., Nat. Rev. Mater. 1 (2016) 16061. doi: 10.1038/natrevmats.2016.61
Y. Omomo, T. Sasaki, L. Wang, et al., J. Am. Chem. Soc. 125 (2003) 3568–3575. doi: 10.1021/ja021364p
J. Cao, F. Wu, M. Wen, et al., Appl. Surf. Sci. 539 (2021) 148164. doi: 10.1016/j.apsusc.2020.148164
L. Zheng, H. Yang, Y. Bai, et al., Energy Mater. Adv. 2022 (2022) 0005. doi: 10.34133/energymatadv.0005
M.H. Alfaruqi, S. Islam, J. Lee, et al., J. Mater. Chem. A 7 (2019) 26966–26974. doi: 10.1039/C9TA09321D
C. Luo, A. Langrock, X. Fan, et al., J. Mater. Chem. A 5 (2017) 18214–18220. doi: 10.1039/C7TA04515H
C. Xie, T. Li, C. Deng, et al., Energy Environ. Sci. 13 (2020) 135–143. doi: 10.1039/C9EE03702K
A. Huang, W. Zhou, A. Wang, et al., Appl. Surf. Sci. 545 (2021) 149041. doi: 10.1016/j.apsusc.2021.149041
L. Zheng, H. Yang, Y. Bai, et al., J. Energy Chem. 60 (2021) 229–232. doi: 10.1016/j.jechem.2020.12.027
L. Zheng, H. Wang, M. Luo, et al., Solid State Ion. 320 (2018) 210–214. doi: 10.1016/j.ssi.2018.02.039

Figure 1 The optimization structure of δ-MnO2. (a) R3M (O3 type). (b) P3M1 (T1 type). (c) P63/mmc (P2 type). (d) C2/m (O'3 type).

Figure 2 The metal ions are adsorbed on the monolayer MnO2. (a-c) Al ions, Mg ions and Na ions on the M-hollow site. (d-f) Al ions, Mg ions and Na ions on the O-hollow site.

Figure 3 The DOS of Na, Mg and Al adsorption on the monolayer MnO2. Due to the valence configurations of Mg-3s and Na-3s, the DOS of Na and Mg are Mg-3s and Na-3s orbitals. The PDOS of Al are showed in Fig. S3 (Supporting information).

Figure 4 The optimal structure of one Al insertion into δ-MnO2. (a) O3 type, (b) T1 type, (c) P2 type, (d) O'3 type.

Figure 5 The density of states of Al insertion into four types of δ-MnO2. (a) O3 type, (b) T1 type, (c) P2 type, (d) O'3 type. And the O3, T1 and O'3 have four different concentrations of Al0.333MnO2, Al0.222MnO2, Al0.111MnO2 and Al0MnO2. The P2 type has five different concentrations of Al0.333MnO2, Al0.25MnO2, Al0.167MnO2, Al0.083MnO2 and Al0MnO2.

Figure 6 The energy barriers of Al ions diffusion in δ-MnO2 of four types. (a) O3 type, (b) T1 type, (c) P2 type, (d) O'3 type.

Figure 7 The initial structure, transition structures and final structure of Al3+ diffusion in the δ-MnO2. (a) O3 type, (b) T1 type, (c) P2 type, (d) O'3 type.

Figure 8 (a) The energy barrier of Al ions diffusion in the P2 type, which the Mn3+ is far away from Al3+. (b) The density of states of Al insertion into MnO2 in the large interlayer spacing of 6.5 Å. (c) The initial structure and transition structure during the Al diffusion in the large interlayer spacing of 6.5 Å. (d) The energy barriers of Al ions diffusion on the monolayer MnO2 and the large interlayer spacing of 6.5 Å.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:



 下载:
下载:
 下载:
下载: