Scheme 1.
Asymmetric hydrofunctionalizations of dienes and our work.
Enantioselective and stereodivergent hydromonofluoroalkylation of conjugated and remote dienes
Qi-Ying Liao , Chao Ma , Yu-Chao Wang , Shao-Qian Yang , Jiang-Shan Ma , Zhi-Tao He

Due to the high electronegativity, lipophilicity and inertness of fluorine atom, the incorporation of a F-containing unit into a target compound represents a privileged and popular strategy in bioactive molecule design, which is corroborated by the statistics that approximately 30%−50% of new pharmaceuticals and agrochemicals contain at least one F atom [1–5]. In especial, alkyl fluorides with a fluorine atom attached to or vicinal to a sp3 carbon stereocenter are known to be very valuable (Scheme 1a) [6–10], because such a unit is considered to enhance three-dimensional conformation, inhibit metabolic decomposition and elevate solubility and bioavailability of a target molecule [1–5]. Despite related studies have been widely carried out, stereoselective construction of alkyl fluorides are still largely unaddressed [11]. Thus, facile protocols for enantioselective preparations of such sites, and even stereodivergent synthesis of all the F-containing isomers, are undoubtedly challenging yet desirable.
As a type of petroleum feedstocks and readily available substrates, diene-involved stereoselective transformations have attracted enormous interests. Among them, transition-metal-catalyzed enantioselective hydrofunctionalization of dienes via π-ƞ3-allylmetal species has emerged as an efficient and atom-economic pathway to build a variety of stereogenic centers [12–24]. Nucleophiles such as amines, N-heteroarenes, stabilized carbons, thiols, sulfinic acids, phosphine oxides, alcohols, oximes etc. have been demonstrated to be suitable for corresponding reactions [25–47]. However, the asymmetric hydromonofluoroalkylation of dienes as an efficient route to stereoselective preparation of alkyl fluorides remains undeveloped (Scheme 1b), presumably due to the reduced stability and nucleophilicity and also the increased steric hindrance of F-containing carbon nucleophiles [48]. The more challenging diastereoselective process with a prochiral fluorinated nucleophile is similarly unexplored. On the other hand, it is a known fact that precise achievement of each stereoisomer of a candidate molecule is crucial in medicinal chemistry, because different isomers might exhibit distinctive pharmacologic effects [49]. Stereodivergent synthesis via synergistic catalysis have provided an efficient solution to access all of stereoisomers of a compound. Since the pioneering work from Carreira, great advance has been achieved from a series of groups, such as Carreira, Zhang, Hartwig, Dong, Wang, Zi, Xiao, Glorious, Lee, He, Gong, Guo et al. [17,34,50–75]. Therefore, the development of stereodivergent hydromonofluoroalkylation of dienes to access all four F-containing isomers is undoubtedly important but has received no study. Finally, asymmetric allylic C—H functionalization is a straightforward route to create stereogenic centers [76–86]. New protocol to achieve allylic monofluoroalkylation via asymmetric allylic C—H functionalization is similarly of high value but has never been explored (Scheme 1b).
Herein, we provide multidimensional solutions to aforementioned longstanding issues (Scheme 1c). First, we describe Pd-catalyzed asymmetric hydromonofluoroalkylation of mono- and disubstituted 1,3-dienes, albeit very recently, an elegant Ni-catalyzed asymmetric process of monosubstituted 1,3-dienes [87] was reported in the course of our submission preparation. Simultaneously, stereodivergent hydromonofluoroalkylation of conjugated dienes for the construction of all four stereoisomers of moieties bearing quaternary F-containing center and vicinal carbon center are also established via synergistic Pd/Cu catalysis. The present protocol is further extended to the realization of asymmetric migratory hydromonofluoroalkylation with skipped dienes. All these three types of transformations, to the best of our knowledge, have not been reported.
We initiated the hydromonofluoroalkylation with conjugated diene 1a as the electrophile and diethyl 2-fluoromalonate 2a as pronucleophile under palladium catalyst. A series of chiral ligands were first evaluated (Scheme 2). As JosiPhos-type ligands performed efficiently in prior asymmetric hydrofunctionalization processes [20,34], L1 did facilitate the generation of 3a, albeit in a low yield and moderate enantioselectivity (entry 1). When an electron-rich ligand L2 was used, the enantioselectivity was elevated to 94% but the reactivity was still low (entry 2). Other JosiPhos-type ligands including L3-L5 and L8-L9 failed to provide promising stereocontrol (entries 3–5, 8 and 9). However, when L6 or L7 was evaluated, 3a was observed in good yields and about 80% ee (entries 6 and 7). We also turned to checking more general biaryl-derived bisphosphine ligands (entries 10–13). Unfortunately, both the efficiency and enantiocontrol of corresponding hydromonofluoroalkylation process decreased heavily. By using Et3N as the solvent, 90% yield and 93% ee of 3a were obtained with L6, while L7 delivered 3a in a slightly eroded enantioselectivity under similar conditions (entries 14 and 15). Finally, with 1.1 equiv. of 1a and 1.0 equiv. of 2a adopted, the transformation proceeded smoothly and provided 3a in 92% isolated yield and 94% ee, and no regioisomer was observed (entry 16). Thus, the optimal conditions for asymmetric hydromonofluoroalkylation were determined as the combination of 1a (1.1 equiv.), 2a (1.0 equiv.) and [Pd]/L6/NaBAr4F (5 mol%) in Et3N at 60 ℃ for a suitable reaction time.

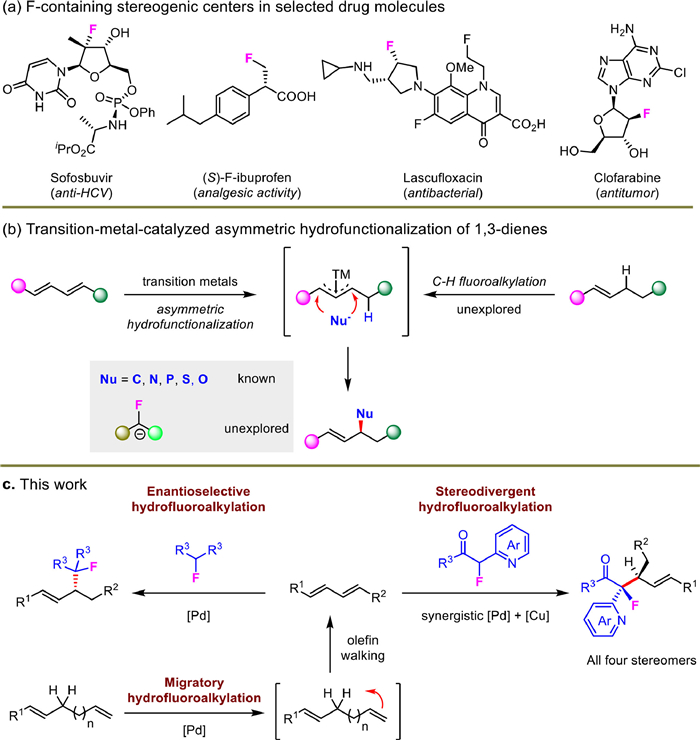
We first checked the substrate generality of conjugated dienes and the results were summarized in Scheme 3. Various functional groups located at different positions of aryl units in diene substrates showed high reaction tolerance (Scheme 3a). For example, aryl-substituted dienes bearing electron-rich alkoxyl, tertiary amine, silyl and OTBS groups or electron-withdrawing CF3, halide and ester units underwent the asymmetric hydrofluoroalkylation in exclusively > 20:1 rr, 87%−99% yields and 93%−95% ee (3a-3i). Heteroaryl such as furan and thiophene-derived dienes were also suitable for this transformation and delivered products 3j and 3k in similarly high efficiency and stereocontrol. When other fluorinated prochiral nucleophiles were adopted instead of 2a, the corresponding products 3l and 3m were prepared in excellent regioselectivities, high yields and enantioselectivities. The structure of 3m was elucidated by X-ray crystal analysis.
Compared with monosubstituted 1,3-dienes, internal dienes are more challenging substrates and typically not included in most of transition-metal-catalyzed hydrofunctionalizations [12,13]. The present hydromonofluoroalkylation protocol was smoothly extended to cover various internal dienes bearing versatile ester units as electrophiles, which greatly broadened the substrate scope (Scheme 3b). For example, esters with alkyl or silyl groups in internal dienes tolerated well with the asymmetric transformation, and good yields and stereocontrol were thus provided (3n-3p). Natural products including nopol, amino alcohol and citronellol-derived dienes were also suitable substrates for present reactions and delivered corresponding hydromonofluoroalkylation products in 10:1–17:1 rr, 63%−93% yield and > 20:1 dr (3q-3s). This route also represents a unique umpolung 1,5-conjugate addition process with challenging fluorinated carbon nucleophiles [45].
Undoubtedly, the ideal goal of asymmetric catalysis is the convenient access to all stereoisomers of a target molecule simultaneously under similar reaction conditions. This is true especially when a tertiary F-containing stereocenter exists, considering that the construction of a fully-substituted enantioenriched fluoride is viewed as the most synthetically challenging process [88]. After the establishment of enantioselective hydromonofluoroalkylation of dienes, an unprecedented stereodivergent coupling of dienes and F-substituted prochiral nucleophiles were developed, inspired by the pioneering work involving Pd-catalyzed stereodivergent hydrofunctionalization by Zi et al. (Scheme 4) [34]. A variety of moieties bearing a fully-substituted fluoride and tertiary carbon stereocenter were constructed in high efficiency and stereoselectivity (Scheme 4a). When 1,3-dienes containing functional groups such as CF3, F, ester, OTBS, Cl, etc. were used, the hydromonofluoroalkylation proceeded in up to 99% yield, > 20:1 dr and > 99% ee (5a-5g). A multi-substituted aryl diene also underwent the transformation in 98% yield, 11:1 dr and 98% ee (5h). The absolute configuration of compound 5c was uncovered via single crystal X-ray diffraction analysis. On the other hand, a couple of fluoroesters bearing pyridine, pyrazine and quinoline units as nucleophiles showed good compatibility with present protocol (5i-5k). α-Fluoroketone also worked as a suitable nucleophile for the diastereoselective hydromonofluoroalkylation (5l). Finally, stereodivergent synthesis all four stereoisomers of 5c was carried out to show the power of present methodology (Scheme 4b). With L17 instead of L16 as the chiral ligand for palladium catalyst, the diastereoisomer of (S,R)−5c, i.e., (S,S)−5c was furnished in excellent yield, enantioselectivity and reasonable diastereoselectivity. The other two stereoisomers were similarly constructed in high efficiency and stereocontrol by simply adjusting the absolute configuration of corresponding chiral ligands.

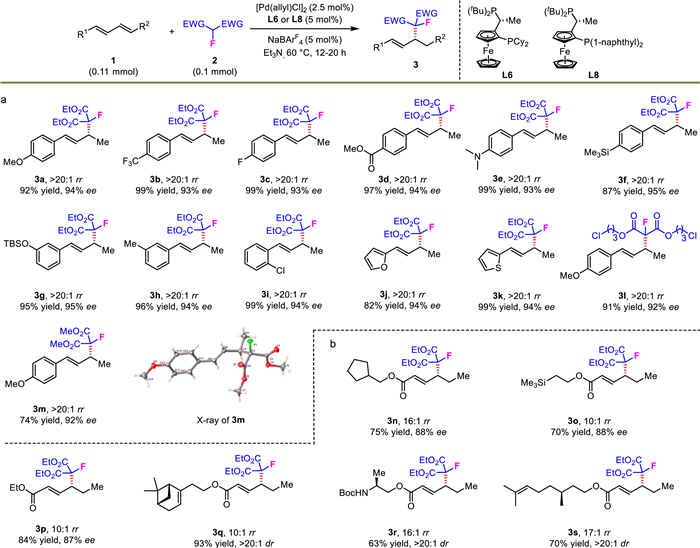
Comparing with traditional allylic monofluoroalkylations with the requirement of prestored leaving groups in the substrates [11,89–92], asymmetric allylic C—H functionalization represents a more straightforward and efficient pathway. Due to the absence of this type of valuable transformation, we moved on to solve this enduring challenge through the development of stereoselective migratory hydromonofluoroalkylation of remote dienes. With the use of 10 mol% Pd(MeCN)4(BF4)2 as palladium source under extended reaction time, the novel migratory hydromonofluoroalkylation reaction performed well (Scheme 5). Diverse skipped dienes 6 as electrophiles reacted with 2a in high stereoselectivity. For example, aryl groups containing alkyl, ether and CF3 units as substituents of skipped dienes were suitable for the migratory coupling (7a-7e). Naphthyl and heteroaryl-derived 1,5-dienes also worked smoothly, furnishing corresponding products in 62%−66% yields and 92%−93% ee (7f-7g). However, when a skipped diene possessing a longer carbon chain between the two olefin units was used, the migratory hydrofunctionalization proceeded in an eroded yield and enantioselectivity (7h). These products above were generally observed with over 20:1 regioselectivity.
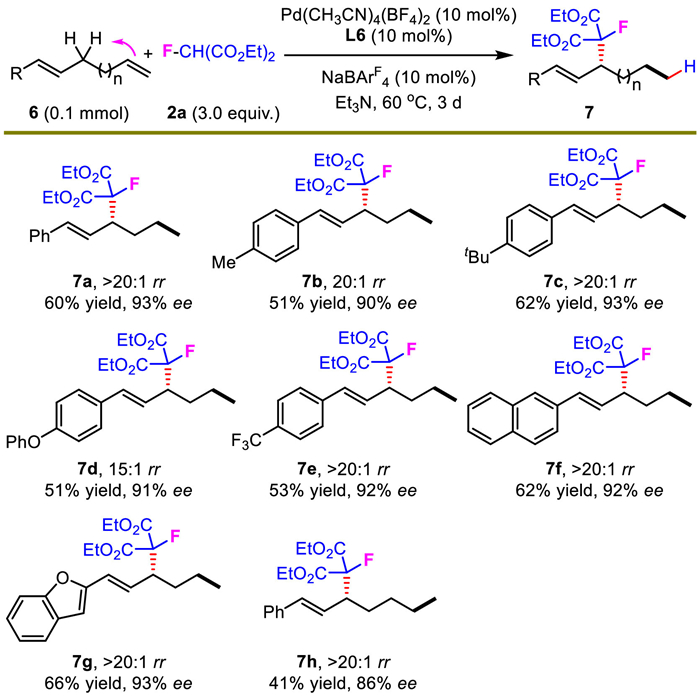
The reliability of the developed hydromonofluoroalkylation was elaborated by a gram-scale test. When 11 mmol of 1a was utilized, 2.6 g of 3m was prepared in 84% yield and 93% ee (Scheme 6). The synthetic value of present methodology was further highlighted by the readily library building of optically active cyclic fluorides. A series of enantioenriched fluorocycles derived from thiourea, boronic acid, carbonate, carbonothioate, sulfite, ketal and phosphonate were constructed with consistently high levels of enantioselectivity (8, 10–12, 14–16). An intriguing fluorinated spirocycle 17 was achieved conveniently from the diol intermediate 9 in 95% yield and 92% ee. In addition, valuable F-substituted small rings, such as oxetane, azetidine and thietane skeletons (13, 19, 20) were also prepared easily in 1–2 steps from 9. All these structurally diverse enantioenriched fluorocycles might provide new chemical space to medicinal chemistry, considering the abundance of fluorine atom, heterocycles and small rings in bioactive molecules.
In conclusion, we have provided a feasible protocol for palladium-catalyzed enantioselective hydromonofluoroalkylation of monosubstituted and internal conjugated dienes. A series of optically active alkyl fluorides are constructed in high yields, regio- and enantioselectivities. Stereodivergent hydromonofluoroalkylation of 1,3-dienes via Pd/Cu dual catalysis is also developed to access all four stereoisomers of alkyl fluorides bearing two vicinal stereogenic centers. Moreover, asymmetric migratory hydromonofluoroalkylation of skipped dienes is established to achieve the straightforward allylic C—H fluoroalkylation. Diverse transformations to build a small library of enantioenriched fluorinated rings show the application potential of these novel hydromonofluoroalkylation methodologies.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge the National Natural Science Foundation of China (NSFC, No. 22071262), Science and Technology Commission of Shanghai Municipality (No. 22ZR1475200), Shanghai RisingStar program (No. 20QA1411300), CAS Key Laboratory of Synthetic Chemistry of Natural Substances, and Shanghai Institute of Organic Chemistry for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
I.M. Jacobson, et al., N. Engl. J. Med. 368 (2013) 1867–1877. doi: 10.1056/NEJMoa1214854
H. Su, Y. Xie, W. Liu, S. You, Bioorg. Med. Chem. Lett. 21 (2011) 3578–3582. doi: 10.1016/j.bmcl.2011.04.114
K. Tanaka, H. Vu, M. Hayashi, J. Infect. Chemother. 27 (2021) 1265–1269. doi: 10.1016/j.jiac.2021.03.026
H.M. Kantarjian, S. Jeha, V. Gandhi, M. Wess, S. Faderl, Leuk. Lymphoma. 48 (2007) 1922–1930. doi: 10.1080/10428190701545644
S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320–330. doi: 10.1039/B610213C
J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432–2506. doi: 10.1021/cr4002879
Y. Zhou, J. Wang, Z. Gu, et al., Chem. Rev. 116 (2016) 422–518. doi: 10.1021/acs.chemrev.5b00392
M. Inoue, Y. Sumii, N. Shibata, ACS Omega 5 (2020) 10633–10640. doi: 10.1021/acsomega.0c00830
Q. Wang, H. Song, Q. Wang, Chin. Chem. Lett. 33 (2022) 626–642. doi: 10.1016/j.cclet.2021.07.064
J. He, Z. Li, G. Dhawan, et al., Chin. Chem. Lett. 34 (2023) 107578. doi: 10.1016/j.cclet.2022.06.001
T.W. Butcher, W.M. Amberg, J.F. Hartwig, Angew. Chem. Int. Ed. 61 (2022) e202112251. doi: 10.1002/anie.202112251
G. Li, X. Huo, X. Jiang, W. Zhang, Chem. Soc. Rev. 49 (2020) 2060–2118. doi: 10.1039/c9cs00400a
N.J. Adamson, S.J. Malcolmson, ACS Catal. 10 (2020) 1060–1076. doi: 10.1021/acscatal.9b04712
H. Zhou, Y. Wang, L. Zhang, M. Cai, S. Luo, J. Am. Chem. Soc. 139 (2017) 3631–3634. doi: 10.1021/jacs.7b00437
X. Wang, B. Wang, X. Yin, et al., Angew. Chem. Int. Ed. 58 (2019) 12264–12270. doi: 10.1002/anie.201905905
Y.H. Yao, H.Y. Yang, M. Chen, et al., J. Am. Chem. Soc. 143 (2021) 85–91. doi: 10.1021/jacs.0c11249
S.Q. Yang, Y.F. Wang, W.C. Zhao, G.Q. Lin, Z.T. He, J. Am. Chem. Soc. 143 (2021) 7285–7291. doi: 10.1021/jacs.1c03157
L. Li, S. Wang, P. Luo, et al., Nat. Commun. 12 (2021) 5667. doi: 10.1038/s41467-021-25981-x
J. Zhang, X. Huo, J. Xiao, et al., J. Am. Chem. Soc. 143 (2021) 12622–12632. doi: 10.1021/jacs.1c05087
Y.W. Chen, Y. Liu, H.Y. Lu, G.Q. Lin, Z.T. He, Nat. Commun. 12 (2021) 5626. doi: 10.1038/s41467-021-25978-6
Z. Yang, J. Wang, Angew. Chem. Int. Ed. 60 (2021) 27288–27292. doi: 10.1002/anie.202112285
S.V. Sieger, I. Lubins, B. Breit, ACS Catal. 12 (2022) 11301–11305. doi: 10.1021/acscatal.2c03105
Q. Li, X. Fang, R. Pan, H. Yao, A. Lin, J. Am. Chem. Soc. 144 (2022) 11364–11376. doi: 10.1021/jacs.2c03620
H.C. Lin, G.J. Knox, C.M. Pearson, et al., Angew. Chem. Int. Ed. 61 (2022) e202201753. doi: 10.1002/anie.202201753
O. Löber, M. Kawatsura, J.F. Hartwig, J. Am. Chem. Soc. 123 (2001) 4366–4367. doi: 10.1021/ja005881o
A. Leitner, J. Larsen, C. Steffens, J.F. Hartwig, J. Org. Chem. 69 (2004) 7552–7557. doi: 10.1021/jo0490999
J. Wilting, M. Janssen, C. Müller, D. Vogt, J. Am. Chem. Soc. 128 (2006) 11374–11375. doi: 10.1021/ja064378u
N.J. Adamson, E. Hull, S.J. Malcolmson, J. Am. Chem. Soc. 139 (2017) 7180–7183. doi: 10.1021/jacs.7b03480
J.S. Marcum, C.C. Roberts, R.S. Manan, T.N. Cervarich, S.J. Meek, J. Am. Chem. Soc. 139 (2017) 15580–15583. doi: 10.1021/jacs.7b08575
N.J. Adamson, K.C.E. Wilbur, S.J. Malcolmson, J. Am. Chem. Soc. 140 (2018) 2761–2764. doi: 10.1021/jacs.7b13300
X.H. Yang, R.T. Davison, V.M. Dong, J. Am. Chem. Soc. 140 (2018) 10443–10446. doi: 10.1021/jacs.8b06957
L. Cheng, M.M. Li, L.J. Xiao, J.H. Xie, Q.L. Zhou, J. Am. Chem. Soc. 140 (2018) 11627–11630. doi: 10.1021/jacs.8b09346
S.Z. Nie, R.T. Davison, V.M. Dong, J. Am. Chem. Soc. 140 (2018) 16450–16454. doi: 10.1021/jacs.8b11150
Q. Zhang, H. Yu, L. Shen, et al., J. Am. Chem. Soc. 141 (2019) 14554–14559. doi: 10.1021/jacs.9b07600
G. Tran, W. Shao, C. Mazet, J. Am. Chem. Soc. 141 (2019) 14814–14822. doi: 10.1021/jacs.9b07253
S. Park, N.J. Adamson, S.J. Malcolmson, Chem. Sci. 10 (2019) 5176–5182. doi: 10.1039/c9sc00633h
Z. Zhang, F. Xiao, H.M. Wu, X.Q. Dong, C.J. Wang, Org. Lett. 22 (2020) 569–574. doi: 10.1021/acs.orglett.9b04341
H. Yang, D. Xing, Chem. Commun. 56 (2020) 3721–3724. doi: 10.1039/d0cc00265h
Q. Zhang, D. Dong, W. Zi, J. Am. Chem. Soc. 142 (2020) 15860–15869. doi: 10.1021/jacs.0c05976
W. Shao, C. Besnard, L. Guénée, C. Mazet, J. Am. Chem. Soc. 142 (2020) 16486–16492. doi: 10.1021/jacs.0c08319
M.M. Li, L. Cheng, L.J. Xiao, J.H. Xie, Q.L. Zhou, Angew. Chem. Int. Ed. 60 (2021) 2948–2951. doi: 10.1002/anie.202012485
J. Xia, T. Hirai, S. Katayama, et al., ACS Catal. 11 (2021) 6643–6655. doi: 10.1021/acscatal.1c01626
A.Y. Jiu, H.S. Slocumb, C.S. Yeung, X.H. Yang, V.M. Dong, Angew. Chem. Int. Ed. 60 (2021) 19660–19664. doi: 10.1002/anie.202105679
J. Long, Y. Li, W. Zhao, G. Yin, Chem. Sci. 13 (2022) 1390–1397. doi: 10.1039/d1sc05651d
Y.C. Wang, Z.X. Xiao, M. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202215568. doi: 10.1002/anie.202215568
S.Q. Yang, A.J. Han, Y. Liu, et al., J. Am. Chem. Soc. 145 (2023) 3915–3925. doi: 10.1021/jacs.2c11843
Q. Li, Z. Wang, V.M. Dong, X.H. Yang, J. Am. Chem. Soc. 145 (2023) 3909–3914. doi: 10.1021/jacs.2c12779
Z. Jiao, J.J. Beiger, Y. Jin, et al., J. Am. Chem. Soc. 138 (2016) 15980–15986. doi: 10.1021/jacs.6b09580
K.M. Rentsch, J. Biochem. Biophys. Methods 54 (2002) 1–9. doi: 10.1016/S0165-022X(02)00124-0
S. Krautwald, E.M. Carreira, J. Am. Chem. Soc. 139 (2017) 5627–5639. doi: 10.1021/jacs.6b13340
I.P. Beletskaya, C. Najera, M. Yus, Chem. Rev. 118 (2018) 5080–5200. doi: 10.1021/acs.chemrev.7b00561
S.J. Kalita, Y.Y. Huang, U. Schneider, Sci. Bull. 65 (2020) 1865–1868. doi: 10.1016/j.scib.2020.08.005
X. Huo, G. Li, X. Wang, W. Zhang, Angew. Chem. Int. Ed. 61 (2022) e202210086. doi: 10.1002/anie.202210086
S. Krautwald, D. Sarlah, M.A. Schafroth, E.M. Carreira, Science 340 (2013) 1065–1068. doi: 10.1126/science.1237068
X. Huo, R. He, X. Zhang, W. Zhang, J. Am. Chem. Soc. 138 (2016) 11093–11096. doi: 10.1021/jacs.6b06156
X. Jiang, J.J. Beiger, J.F. Hartwig, J. Am. Chem. Soc. 139 (2017) 87–90. doi: 10.1021/jacs.6b11692
F.A. Cruz, V.M. Dong, J. Am. Chem. Soc. 139 (2017) 1029–1032. doi: 10.1021/jacs.6b10680
L. Wei, Q. Zhu, S.M. Xu, X. Chang, C.J. Wang, J. Am. Chem. Soc. 140 (2018) 1508–1513. doi: 10.1021/jacs.7b12174
Z.T. He, X. Jiang, J.F. Hartwig, J. Am. Chem. Soc. 141 (2019) 13066–13073. doi: 10.1021/jacs.9b04440
M.M. Zhang, Y.N. Wang, B.C. Wang, et al., Nat. Commun. 10 (2019) 2716. doi: 10.1038/s41467-019-10674-3
S. Singha, E. Serrano, S. Mondal, C.G. Daniliuc, F. Glorius, Nat. Catal. 3 (2020) 48–54.
R. He, X. Huo, L. Zhao, et al., J. Am. Chem. Soc. 142 (2020) 8097–8103. doi: 10.1021/jacs.0c02150
Y. Peng, X. Huo, Y. Luo, L. Wu, W. Zhang, Angew. Chem. Int. Ed. 60 (2021) 24941–24949. doi: 10.1002/anie.202111842
B. Kim, Y. Kim, S.Y. Lee, J. Am. Chem. Soc. 143 (2021) 73–79. doi: 10.1021/jacs.0c11077
H. Wang, R. Zhang, Q. Zhang, W. Zi, J. Am. Chem. Soc. 143 (2021) 10948–10962. doi: 10.1021/jacs.1c02220
Q. Zhang, M. Zhu, W. Zi, Chem 8 (2022) 2784–2796. doi: 10.1016/j.chempr.2022.07.014
Y.H. Wen, Z.J. Zhang, S. Li, J. Song, L.Z. Gong, Nat. Commun. 13 (2022) 1344. doi: 10.1038/s41467-022-29059-0
Q. Hu, Z. He, L. Peng, C. Guo, Nat. Synth. 1 (2022) 322–331. doi: 10.1038/s44160-022-00050-3
Z. He, L. Peng, C. Guo, Nat. Synth. 1 (2022) 393–400. doi: 10.1038/s44160-022-00063-y
T.P. Le, S. Tanaka, M. Yoshimura, K. Sato, M. Kitamura, Nat. Commun. 13 (2022) 5876. doi: 10.1038/s41467-022-33432-4
L. Xiao, X. Chang, H. Xu, et al., Angew. Chem. Int. Ed. 61 (2022) e202212948. doi: 10.1002/anie.202212948
X. Chang, X. Cheng, X.T. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202206517. doi: 10.1002/anie.202206517
J. Han, R. Liu, Z. Lin, W. Zi, Angew. Chem. Int. Ed. 61 (2022) e202215714.
M. Zhu, Q. Zhang, W. Zi, Angew. Chem. Int. Ed. 60 (2022) 6545. doi: 10.3390/app12136545
W. Chai, B. Guo, Q. Zhang, W. Zi, Chem. Catal. 2 (2022) 1428. doi: 10.1016/j.checat.2022.04.006
R. Wang, Y. Luan, M. Ye, Chin. J. Chem. 37 (2019) 720–743. doi: 10.1002/cjoc.201900140
P.S. Wang, L.Z. Gong, Acc. Chem. Res. 53 (2020) 2841–2854. doi: 10.1021/acs.accounts.0c00477
D.J. Covell, M.C. White, Angew. Chem. Int. Ed. 47 (2008) 6448–6451. doi: 10.1002/anie.200802106
H. Du, B. Zhao, Y. Shi, J. Am. Chem. Soc. 130 (2008) 8590–8591. doi: 10.1021/ja8027394
Z. Chai, T.J. Rainey, J. Am. Chem. Soc. 134 (2012) 3615–3618. doi: 10.1021/ja2102407
P.S. Wang, H.C. Lin, Y.J. Zhai, Z.Y. Han, L.Z. Gong, Angew. Chem. Int. Ed. 53 (2014) 12218–12221. doi: 10.1002/anie.201408199
B.M. Trost, E.J. Donckele, D.A. Thaisrivongs, M. Osipov, J.T. Masters, J. Am. Chem. Soc. 137 (2015) 2776–2784. doi: 10.1021/jacs.5b00786
W. Liu, S.Z. Ali, S.E. Ammann, M.C. White, J. Am. Chem. Soc. 140 (2018) 10658–10662. doi: 10.1021/jacs.8b05668
T.C. Wang, L.F. Fan, Y. Shen, P.S. Wang, L.Z. Gong, J. Am. Chem. Soc. 141 (2019) 10616–10620. doi: 10.1021/jacs.9b05247
R. Yu, R. Shanmugam, X. Fang, Angew. Chem. Int. Ed. 59 (2020) 21436–21441. doi: 10.1002/anie.202008854
Y. Bunno, Y. Tsukimawashi, M. Kojima, T. Yoshino, S. Matsunaga, ACS Catal. 11 (2021) 2663–2668. doi: 10.1021/acscatal.0c05261
L. Liao, Y. Zhang, Z. Wu, et al., J. Yu, Chem. Sci. 13 (2022) 12519–12526. doi: 10.1039/d2sc03958c
Y. Zhu, J. Han, J. Wang, et al., Chem. Rev. 118 (2018) 3887–3964. doi: 10.1021/acs.chemrev.7b00778
E. Belanger, K. Cantin, O. Messe, M. Tremblay, J.F. Paquin, J. Am. Chem. Soc. 129 (2007) 1034–1035. doi: 10.1021/ja067501q
T. Song, X. Zhao, J. Hu, W. Dan, Eur. J. Org. Chem. 2018 (2018) 1141–1144. doi: 10.1002/ejoc.201701739
A. Sripada, C. Wolf, J. Org. Chem. 87 (2022) 11880–11887. doi: 10.1021/acs.joc.2c01414
J. Han, L. Hoteite, J.P.A. Harrity, Chem. Eur. J. 28 (2022) e202201595. doi: 10.1002/chem.202201595

Scheme 2 Reaction development. The yield was determined by 1H NMR and the ee was determined by HPLC analysis. a 2a (1.5 equiv.) and tBuONa (10 mol%) were used. b Et3N as the solvent. c 1a (1.1 equiv.) and 2a (1.0 equiv.) were used. d Isolated yield.

Scheme 3 Enantioselective hydromonofluoroalkylation of conjugated dienes. Isolated yield. The ee was determined by HPLC analysis. For 3a-3m, L6 was used. For 3n-3s, 1 (1.0 equiv.), 2 (1.5 equiv.), [Pd(allyl)Cl]2 (2.5 mol%), L8 (5 mol%) as the chiral ligand, NaBAr4F (5 mol%, ArF = 3,5-(CF3)2C6H3), Et3N (3.0 equiv.) as the base and tBuONa (10 mol%) as additives in PhCF3 at 60 ℃ for 12–20 h were adopted as the standard reaction conditions.

Scheme 4 Stereodivergent hydromonofluoroalkylation of 1,3-dienes. Isolated yield. The rr and dr values were determined by 1H NMR. The ee was determined by HPLC analysis. a (S)-L16 and (S,S)-Ph-BPE were used. b DBU (5 mol%) was used. c Toluene as the solvent at −10 ℃ to 0 ℃ were adopted.

Scheme 5 Migratory hydrofluoroalkylation of 1,3-dienes. The rr values were determined by 19F NMR. The ee was determined by HPLC analysis. Isolated yields.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
