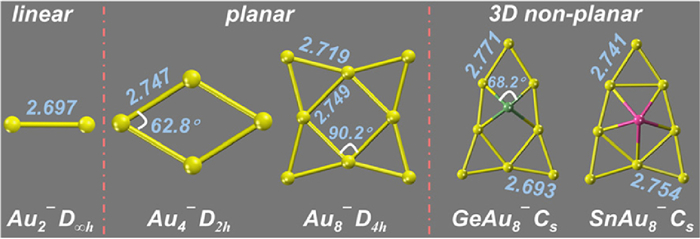
Figure 1.
Calculated lowest-energy structures of Aun¯ (n = 2, 4 and 8) and MAu8¯ (M = Ge, Sn). Selected bond lengths and angles are indicated with units of angstrom (Å) and angle (°).
Solvent field regulated superhalogen in pure and doped gold cluster anions
Hao Wang , Jun Li , Jing Chen , Yuxiang Bu , Shi-Bo Cheng

Past several decades have witnessed the booming progress in cluster science from both the fundamental theory and experiments [1-7]. Atomic clusters have received increasing attention owing to their fascinating structures and properties together with their potential applications in various disciplines, e.g., catalysis, materials and energy [8-10]. Superatom, a special branch of atomic clusters proposed theoretically by Jena and Khanna [11,12], is designed to mimic properties of single elements in the traditional two-dimensional (2D) periodic table, which has the potential in constructing a new 3D periodic table [3,13,14]. Among numerous superatoms, superhalogen, originally proposed in the pioneering work of Gutsev and Boldyrev [15], is one of the most explored categories because of its simple and well-defined criterion. It was suggested to describe clusters possessing extremely high electron-attaching capability, corresponding to the strong oxidation capability that can even oxidize the noble gas atom [16], (H2O)n clusters [17], etc. It also includes a series of highly stable cluster anions (superhalogen anions) [18,19], e.g., BF4¯, AsF6¯ and AlCl4¯, possessing high vertical detachment energies (VDE), which are larger than that of a halogen atom (3.06–3.62 eV) [1,15,20].
Following the first experimental evidence of the superhalogen from Castleman et al. [1], many research groups contribute to this field and many superhalogens have been identified [21-32]. The earliest construction of superhalogens was carried out by a [MXk+1]−1 formula, where M stands for a central atom with a maximal formal valence k, enclosed by halogen ligands X [15]. Subsequently, different electron-counting rules (ECRs), such as the Jellium model, the 18-electron rule, the Wade-Mingos rule, and Hückel 4n+2 rule [33-37], were successfully developed to design superhalogens. However, there exists obvious limitations in these traditional methodologies because one needs to alter the intrinsic properties, e.g., the composition or the number of valence electrons, of parent clusters to obtain superhalogens, which is relatively difficult and inconvenient to control in experiments. To overcome such a drawback, we developed external-field strategies for designing superhalogens recently. The oriented external electric field (OEEF) and ligand field strategies [20,38-40], which can significantly increase the electron-attaching capability of metal clusters while maintaining their components, the valence shell filling, and the geometrical and energetic stability, were proposed as effective methodologies in constructing superhalogens. Compared with conventional ECRs, these external-field strategies may provide promising avenues in the superhalogen synthesis conveniently and in boosting the potential applications of superatoms.
Additionally, accompanied by the gas-phase cluster research, the condensed-phase atomically precise cluster synthesis has also experienced a rapid development during the past decade [41-44]. In such a process, apart from the ligands that can protect the cluster cores to avoid their aggregation, various solvents also provide a crucial environment to stabilize clusters. Note that, it has been well-established that the solvent, an important medium in the condensed-phase synthesis, can assist in stabilizing unstable anions [45] and intermediates [46] as well as altering the reactivity of certain organic reactions [47]. Its capability in regulating the electronic properties of clusters for the superatom design, however, has not been understood. Additionally, apart from the ligand field and OEEF we investigated, are there any other external field strategies that can work in the superhalogen design? These questions inspire us to examine whether the solvent can work as a novel external field in realizing the superhalogen construction. If so, it will represent a great progress in the superhalogen synthesis since solvent are indispensable, relatively inexpensive, and conveniently available in the cluster synthesis. Thus, in this communication, the density functional theory (DFT), molecular dynamics (MD) simulation, and quantum mechanics/molecular mechanics (QM/MM) simulation were employed to explore the capability of different polar solvents in modulating the electronic properties of typical gold cluster anions to construct novel superhalogens. Such a capability was first evidenced in model gas-phase pure and doped gold clusters, and subsequently extended to a real experimentally synthesized Au18 nanocluster.
We first investigated whether solvents could regulate the electronic properties of naked cluster anions. Several typical anions, i.e., Aun¯ (n = 2, 4 and 8) and MAu8¯ (M = Ge, Sn), were adopted as model systems. It is well-accepted that the superhalogen anion has a unique characteristic with a strong capability of binding an excess electron, corresponding to a high VDE. The global minima of these anions were optimized, which are displayed in Fig. 1. Here, we adopted clusters with various geometries, which are linear, planar, and 3D structures, respectively. This can offer relative comprehensive model systems to understand the regulation effect of solvents regardless of special clusters' geometries. According to the optimized structures, their VDEs were calculated to be about 2.15, 2.74, 2.99, 2.90 and 2.92 eV, respectively, which are all non-superhalogens. Therefore, these anions are good candidates to understand the transformation between non-superhalogens and superhalogen anions under solvents. Next, several commonly used polar solvents, which are toluene, dichloromethane (DCM), methanol, dimethylsulfoxide (DMSO), and water with gradually increasing static dielectric constants (ɛ) of 2.37, 8.93, 32.61, 46.83 and 78.34 (Table S1 in Supporting information), respectively, were employed to examine their regulation effects on the clusters' VDEs. The calculated variation trends of the VDEs of these five anions in different solvents are listed in Fig. 2a and Fig. S1 (Supporting information). Apparently, compared with the cases in the gas phase, the solvent field remarkably increases their VDEs in all model cluster anions. Taking Au4¯ as an example (Fig. 2a), its VDE monotonically increases accompanied by the enhancement of the polarity of solvents. Specifically, the VDE of the gas-phase Au4¯ was estimated to be 2.74 eV, in line with the experimental measurement (2.75 eV) [48]. This finding clearly demonstrates the accuracy and reliability of the present level of theory. And its VDE goes up to 3.18 eV (superhalogen) upon the introduction of toluene. This indicates that such a weak solvent field can even realize the superhalogen conversion from the gas-phase non-superhalogen Au4¯. Subsequently, the water solvent can further enhance the electron-binding capability of Au4¯ to a VDE of 3.50 eV. Similar regulation effect of the solvent field also valid in other model systems (Fig. S1). Therefore, these findings provide solid evidence that the solvent field can be considered as an effective external field in the superhalogen modulation.
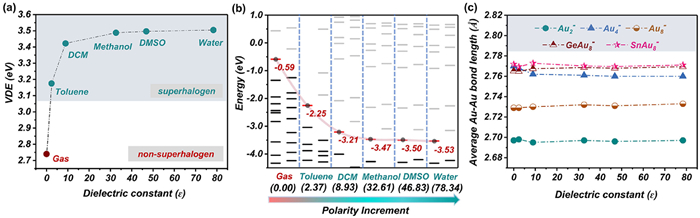
It is well-accepted that the highest occupied molecular orbital (HOMO) of the anion governs the stability of the outmost electron. The lower the HOMO level is, the stronger the ability of the anion to bind the outermost electron. To understand the microscopic origin of the above VDE changes, the one-electron energy levels of cluster anions in different solvents were computed. Taking Au4¯ as an instance again (Fig. 2b), it is remarkable that the introduction of polar solvents significantly decreases the cluster's HOMO levels. To be specific, the HOMO level drops sharply from the original −0.59 V to −2.25 eV when the environment changed from the gas phase to toluene, representing an apparent effect of toluene on the electron-binding ability of Au4¯. Moreover, the HOMO level declines continuously accompanied by the increment of the solvent polarity, implying that the solvent field can stabilize Au4¯ and make it harder to lose the outmost electron. These findings correlate well with the VDE enhancement of Au4¯ in different solvents (Fig. 2a), which demonstrate that the downward shift of the electronic spectrum is probably the origin for the changes from non-superhalogen to superhalogen in different solvents. Having understood the effect of different solvents on the electronic properties of these cluster anions, we want to further examine whether the field alters their geometrical and electronic structures. The average Au-Au bond lengths of the cluster anions were calculated (Fig. 2c). Surprisingly, compared with the situation in the gas phase, the average Au-Au bond lengths almost stay unchanged under different solvent environments. Additionally, the frontier MOs of the model clusters were also evaluated. Taking Au4¯ and Au8¯ as examples (Tables S2 and S3 in Supporting information), their gas-phase electronic structures are well preserved in different solvent fields. All these findings undoubtably unveil that the solvent field can remarkably enhance these anions' electron-binding ability while maintaining their geometrical and electronic stability, exhibiting a good selectivity in the superhalogen design.
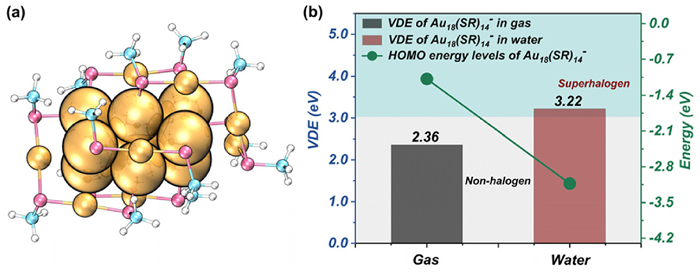
Furthermore, it is necessary to note that naked clusters are highly reactive and easy to coalesce in the solvent environment, and protected ligands are indispensable in the condensed-phase syntheses of clusters. Thus, although the above results reveal the regulation effect of polar solvents in model clusters, exploring its modulation power in practical cluster systems, like the ligand-protected nanoclusters, will deliver more valuable guidance information about the solvent-assisted superhalogen synthesis in the condensed phase. To this end, a stable gold nanocluster, namely Au18(SR)14 (SR = SC6H11), which was experimentally identified in 2015 [49,50], was adopted here. Based on the reported crystal structure of Au18(SR)14 [49,50], we optimized the geometry of Au18(SR)14¯ (Fig. 3a). To simplify the calculations, the C6H11 moiety was replaced by CH3, whose reliability was verified previously [50]. The main objective of this part of calculation is to determine whether the solvent field can also increase the VDE of a practical gold nanocluster, termed as Au18_nc (Au18(SCH3)14) in the following, to realize the superhalogen design. The water solvent was utilized here because it has the largest effect in lifting the VDE of the model anions (Fig. 2a and Fig. S1). The implicit solvation model was first applied. The VDE of gas-phase Au18_nc¯ was computed to be 2.36 eV (Fig. 3b), which is a non-superhalogen. Inspiringly, the cluster's VDE increases to 3.22 eV by simply introducing H2O, reaching the superhalogen zone. This demonstrates that the solvent field is indeed an effective external field that can assist in the superhalogen formation for both the simplified model naked clusters and practical ligand-protected nanoclusters. Inspection of the HOMO levels of Au18_nc¯ in the gas phase (−1.08 eV) and liquid phase (−3.13 eV) also unveils that H2O can significantly stabilize the HOMO level of the nanocluster (Fig. 3b), enhancing the electron-binding capability of the anion and corresponds to the observed VDE increment in H2O. Additionally, like the cases in the model anions, the H2O solvent has little effect on the structure of Au18_nc¯ (Table S4 in Supporting information). Therefore, it is reasonable to suggest that, apart from the naked cluster anions, the solvent field can also regulate the electronic properties of practical nanoclusters forming superhalogens without destroying their structural stability, which may boost the potential applications of these superhalogens because ligands are crucial in the condensed-phase synthesis of nanoclusters.
In addition, it has been well-established that the water solvent itself has a strong power to stabilize one extra electron forming the solvated electron [51-53]. For instance, by using the magnetic bottle photoelectron technique, a VDE of 3.7 ± 0.1 eV for the bulk water was obtained experimentally [52]. Thus, one important question is that does the VDE increment observed above stem from the solvent itself or the solvent-assisted nanocluster? To address this puzzle, two different models were conducted, which are the DFT calculations with the explicit solvation model and the QM/MM simulations, respectively. Here, 82 H2O were placed around Au18_nc, termed as Au18_nc@82H2O, followed by the optimization of the whole system (Fig. 4a). The VDE of the optimized Au18_nc@82H2O¯ was calculated to be 3.32 eV (a superhalogen), which agrees well with the value (3.22 eV) obtained in the SMD model (Fig. 3b). More importantly, the spin density calculation on Au18_nc@82H2O¯ obviously reveals that the extra electron almost locates in Au18_nc rather than the surrounding H2O (Fig. 4b), which answers the above question. The NBO charge analysis [54] can also testify such a fact (Table S5 in Supporting information). The central Au18_nc core and the outside H2O gain 0.873 and 0.127 |e|, respectively, when one more electron is introduced into the neutral system. This further demonstrates that the extra electron mainly gathers in Au18_nc. Thus, the solvent field can indeed enhance the electron-binding ability of the nanocluster anion. In addition, in such an explicit solvation model, the geometrical and electronic structures of the central Au18_nc¯ are maintained as well compared with those in the gas phase (Table S6 and Fig. S2 in Supporting information).
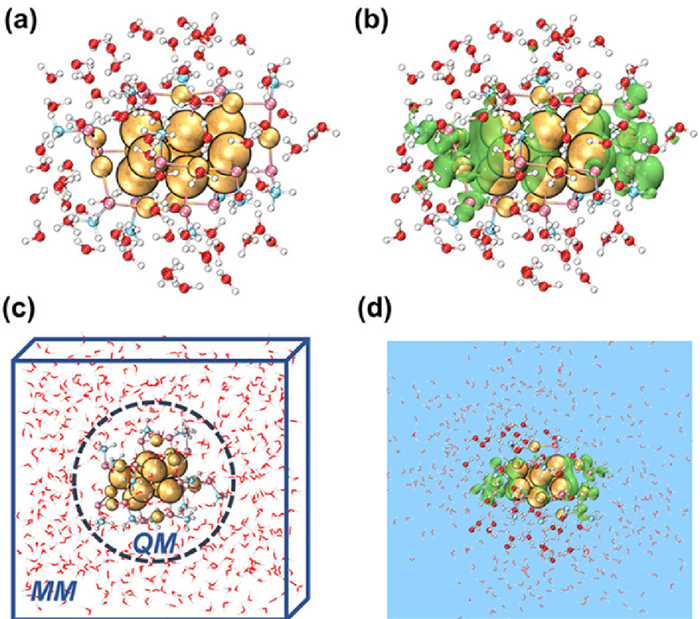
To better understand the real situation of the aqueous solvents around the nanocluster, a QM/MM simulation by including 543 H2O, 43 of which were considered as the QM region, termed as Au18_nc¯@43H2O@500H2O, was performed for the electronic property predictions (Fig. 4c). Fig. S3 (Supporting information) shows the 3 ps simulation to acquire the neutral steady state structure, based on which an electron was vertically added to conduct the following 2 ps QM/MM simulation to understand the anion's dynamic information (Fig. S4a in Supporting information). And Fig. S4 exhibits the anion's energetic and structural stability. Importantly, the electron spin density diagram shows that the extra electron locates around Au18_nc (Fig. 4d), which is consistent with the result obtained from the explicit solvation model (Fig. 4b). Moreover, we also examined the charge variation (ΔQ) for the anionic Au18_nc and surrounding 43 H2O in the QM zone, and it is apparent that the extra electron most locates in the Au18_nc core (Fig. S5 in Supporting information). Consequently, the consistency between the DFT calculations and QM/MM simulation undoubtedly clarify that the solvent field can improve the electron-binding capability of the cluster itself to enable it to possess the superhalogen characteristic.
Thus, above findings show that the solvent field has a dramatic regulation effect on the electronic properties of both the model naked cluster anions and the practical nanocluster producing superhalogens. The significance and advantage of this field lie in solvents are indispensable in the condensed-phase syntheses of atomically precise clusters. Compared with another two external fields, i.e., the ligand field and OEEF reported in our group [20,38-40,55], the solvent field is the easiest one to achieve in experiments, and one can obtain superhalogens by simply altering polar solvents used in the condensed-phase experiments. Strikingly, these findings highlighted here not only show the capability of polar solvents in the superhalogen regulation but also provide another important jigsaw in the whole picture of external-field-regulated superhalogens (EFRS).
In summary, polar solvents were demonstrated to possess the power to modulate the electronic property of cluster anions in constructing the superhalogen anions. Such a regulation effect was observed to work in both the model gas-phase gold clusters and an experimentally synthesized Au18 nanocluster. Different from traditional ECRs, the solvent field, which is a convenient and noninvasive methodology, can be considered as a novel external field to significantly increase the electron-binding capability of cluster anions without disturbing their geometrical and electronic structures. It was also revealed that the striking increment of the electron-binding capability of the anion stems from the downward shift of the cluster's electronic spectrum. Moreover, we also demonstrated that the extra electron mainly locates on the cluster core itself rather than the surrounding solvent molecules. Considering such an external field is an indispensable part currently used in the condensed-phase synthesis of nanoclusters, these findings are promising to boost the potential applications of superatoms, and we wish our results can spur more efforts in experimentally synthesizing various superhalogens in the condensed phase by using suitable solvents.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work is supported by the National Natural Science Foundation of China (NSFC, No. 92161101), the Taishan Scholars Project of Shandong Province (No. ts201712011), the Innovation Project of Jinan Science and Technology Bureau (No. 2021GXRC032). The scientific calculations in this paper have been done on the HPC Cloud Platform of Shandong University.
Supplementary material associated with this article can be found, in the online version, at doi:
D.E. Bergeron, A.W. Castleman Jr., T. Morisato, S.N. Khanna, Science 304 (2004) 84–87. doi: 10.1126/science.1093902
S.B. Cheng, C. Berkdemir, A.W. Castleman Jr., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 4941–4945. doi: 10.1073/pnas.1504714112
R.E. Leuchtner, A.C. Harms, A.W. Castleman Jr., J. Chem. Phys. 91 (1989) 2753–2754. doi: 10.1063/1.456988
Z. Luo, A.W. Castleman Jr., Acc. Chem. Res. 47 (2014) 2931–2940. doi: 10.1021/ar5001583
Q. Du, B. Yin, S. Zhou, Z. Luo, J.J. Zhao, Chin. Chem. Lett. 33 (2022) 995–1000. doi: 10.1016/j.cclet.2021.08.127
L.S. Chen, Y.Z. Liu, X.N. Li, et al., J. Phys. Chem. Lett. 12 (2021) 6519–6525. doi: 10.1021/acs.jpclett.1c01584
S.M. Lang, T.M. Bernhardt, M. Krstić, V. Bonačić-Koutecký, Angew. Chem. Int. Ed. 53 (2014) 5467–5471. doi: 10.1002/anie.201310134
P. Jena, Q. Sun, Chem. Rev. 118 (2018) 5755–5870. doi: 10.1021/acs.chemrev.7b00524
A. Cheng, R. Wang, Z. Liu, et al., J. Phys. Chem. Lett. 12 (2021) 8713–8719. doi: 10.1021/acs.jpclett.1c02439
M. Liu, X. Ren, X. Liu, et al., Chin. Chem. Lett. 31 (2020) 3117–3120. doi: 10.1016/j.cclet.2020.06.024
S.N. Khanna, P. Jena, Phys. Rev. B 51 (1995) 13705–13716. doi: 10.1103/PhysRevB.51.13705
S.N. Khanna, P. Jena, Phys. Rev. Lett. 69 (1992) 1664–1667. doi: 10.1103/PhysRevLett.69.1664
A.W. Castleman Jr., P. Jena, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 10554–10559. doi: 10.1073/pnas.0601780103
T. Inoshita, S. Ohnishi, A. Oshiyama, Phys. Rev. Lett. 57 (1986) 2560–2563. doi: 10.1103/PhysRevLett.57.2560
G.L. Gutsev, A.I. Boldyrev, Chem. Phys. 56 (1981) 277–283. doi: 10.1016/0301-0104(81)80150-4
N. Bartlett, Proc. Chem. Soc. 115 (1962) 218.
M. Marchaj, S. Freza, O. Rybacka, P. Skurski, Chem. Phys. Lett. 574 (2013) 13–17. doi: 10.1016/j.cplett.2013.05.009
I. Anusiewicz, M. Sobczyk, I. D ˛ abkowska, P. Skurski, Chem. Phys. 291 (2003) 171–180. doi: 10.1016/S0301-0104(03)00208-8
P. Koirala, M. Willis, B. Kiran, A.K. Kandalam, P. Jena, J. Phys. Chem. C 114 (2010) 16018–16024. doi: 10.1021/jp101807s
Y. Zhao, J. Wang, H.C. Huang, et al., J. Phys. Chem. Lett. 11 (2020) 1093–1099. doi: 10.1021/acs.jpclett.9b03794
X.B. Wang, C.F. Ding, L.S. Wang, A.I. Boldyrev, J. Simons, J. Chem. Phys. 110 (1999) 4763–4771. doi: 10.1063/1.478386
Y. Gao, S. Bulusu, X.C. Zeng, J. Am. Chem. Soc. 127 (2005) 15680–15681. doi: 10.1021/ja055407o
Y. Gao, H. Banjade, M. Wu, P. Jena, J. Phys. Chem. Lett. 13 (2022) 1049–1056. doi: 10.1021/acs.jpclett.1c04119
A.N. Alexandrova, A.I. Boldyrev, Y.J. Fu, et al., J. Chem. Phys. 121 (2004) 5709–5719. doi: 10.1063/1.1783276
B.M. Elliott, E. Koyle, A.I. Boldyrev, X.B. Wang, L.S. Wang, J. Phys. Chem. A 109 (2005) 11560–11567. doi: 10.1021/jp054036v
V. Chauhan, A.C. Reber, S.N. Khanna, J. Am. Chem. Soc. 139 (2017) 1871–1877. doi: 10.1021/jacs.6b09416
H. Yang, Y. Li, H.M. He, et al., Chem. Phys. Lett. 713 (2018) 203–209. doi: 10.1016/j.cplett.2018.10.039
Z. Liu, X. Liu, J. Zhao, Nanoscale 9 (2017) 18781–18787. doi: 10.1039/C7NR06431D
A.K. Srivastava, N. Misra, Theor. Chem. Acc. 134 (2015) 93. doi: 10.1007/s00214-015-1696-5
M. Czapla, O. Ciepła, J. Brzeski, P. Skurski, J. Phys. Chem. A 122 (2018) 8539–8548. doi: 10.1021/acs.jpca.8b07514
I. Świerszcz, I. Anusiewicz, Mol. Phys. 111 (2013) 377–385. doi: 10.1080/00268976.2012.726378
C. Sikorska, Int. J. Quantum Chem. 118 (2018) e25728.
W.D. Knight, K. Clemenger, W.A. De Heer, et al., Phys. Rev. Lett. 52 (1984) 2141–2143. doi: 10.1103/PhysRevLett.52.2141
H.J. Zhai, J. Li, L.S. Wang, J. Chem. Phys. 121 (2004) 8369–8374. doi: 10.1063/1.1799574
B. Pathak, D. Samanta, R. Ahuja, P. Jena, ChemPhysChem 12 (2011) 2423–2428. doi: 10.1002/cphc.201100320
S. Giri, S. Behera, P. Jena, Angew. Chem. Int. Ed. 53 (2014) 13916–13919. doi: 10.1002/anie.201408648
B.Z. Child, S. Giri, S. Gronert, P. Jena, Chem. Eur. J. 20 (2014) 4736–4745. doi: 10.1002/chem.201305057
Y.J. Duan, Y. Zhao, S.B. Cheng, Q. Wei, J. Phys. Chem. A 126 (2022) 29–35. doi: 10.1021/acs.jpca.1c08452
J. Li, H.C. Huang, J. Wang, et al., Nanoscale 11 (2019) 19903–19911. doi: 10.1039/C9NR05613K
X.X. Dong, Y. Zhao, J. Li, et al., J. Phys. Chem. Lett. 13 (2022) 3942–3948. doi: 10.1021/acs.jpclett.2c00916
T. Omoda, S. Takano, S. Masuda, T. Tsukuda, Chem. Commun. 57 (2021) 12159–12162. doi: 10.1039/D1CC05235G
T. Higaki, Q. Li, M. Zhou, et al., Acc. Chem. Res. 51 (2018) 2764–2773. doi: 10.1021/acs.accounts.8b00383
P. Maity, S. Xie, M. Yamauchi, T. Tsukuda, Nanoscale 4 (2012) 4027–4037. doi: 10.1039/c2nr30900a
S. Takano, T. Tsukuda, J. Am. Chem. Soc. 143 (2021) 1683–1698. doi: 10.1021/jacs.0c11465
H. Zhang, Q. Luo, S. Cheng, Q. Fu, Y. Bu, J. Phys. Chem. C 122 (2018) 28466–28477. doi: 10.1021/acs.jpcc.8b07744
J. Santatiwongchai, K. Faungnawakij, P. Hirunsit, ACS Catal. 11 (2021) 9688–9701. doi: 10.1021/acscatal.1c01486
K. Ueno, Y. Ishimizu, H. Fujii, Inorg. Chem. 60 (2021) 9243–9247. doi: 10.1021/acs.inorgchem.1c01018
H. Häkkinen, B. Yoon, U. Landman, et al., J. Phys. Chem. A 107 (2003) 6168–6175. doi: 10.1021/jp035437i
A. Das, C. Liu, H.Y. Byun, et al., Angew. Chem. Int. Ed. 54 (2015) 3140–3144. doi: 10.1002/anie.201410161
S. Chen, S. Wang, J. Zhong, et al., Angew. Chem. Int. Ed. 54 (2015) 3145–3149. doi: 10.1002/anie.201410295
K. Majer, L. Ma, B. Von Issendorff, J. Phys. Chem. A 125 (2021) 8426–8433. doi: 10.1021/acs.jpca.1c06761
D. Luckhaus, Y.I. Yamamoto, T. Suzuki, R. Signorell, Sci. Adv. 3 (2017) e1603224. doi: 10.1126/sciadv.1603224
Y. Tang, H. Shen, K. Sekiguchi, et al., Phys. Chem. Chem. Phys. 12 (2010) 3653–3655. doi: 10.1039/b925741a
J.P. Foster, F. Weinhold, J. Am. Chem. Soc. 102 (1980) 7211–7218. doi: 10.1021/ja00544a007
J. Li, H. Huang, J. Chen, Y. Bu, S. Cheng, Nano Res. 15 (2022) 1162–1170. doi: 10.1007/s12274-021-3619-1

Figure 1 Calculated lowest-energy structures of Aun¯ (n = 2, 4 and 8) and MAu8¯ (M = Ge, Sn). Selected bond lengths and angles are indicated with units of angstrom (Å) and angle (°).

Figure 2 (a) Theoretical VDE values (the lower limit of the superhalogen (3.06 eV for the I atom) is indicated) and (b) one-electron energy levels (red lines signify the HOMO levels, black lines represent other occupied MOs and gray lines designate the unoccupied MOs) of Au4¯ together with (c) the average Au-Au bond lengths (in Å) of Aun¯ (n = 2, 4 and 8) and MAu8¯ (M = Ge, Sn) in the gas phase and different solvents.

Figure 3 (a) The optimized geometry of the Au18(SCH3)14¯ cluster in water and (b) calculated VDE values and HOMO levels of Au18(SCH3)14¯ in the gas phase and water, respectively. In (a), all Au atoms are identical, and the central nine gold atoms are enlarged in size to just highlight the Au9 kernel.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
