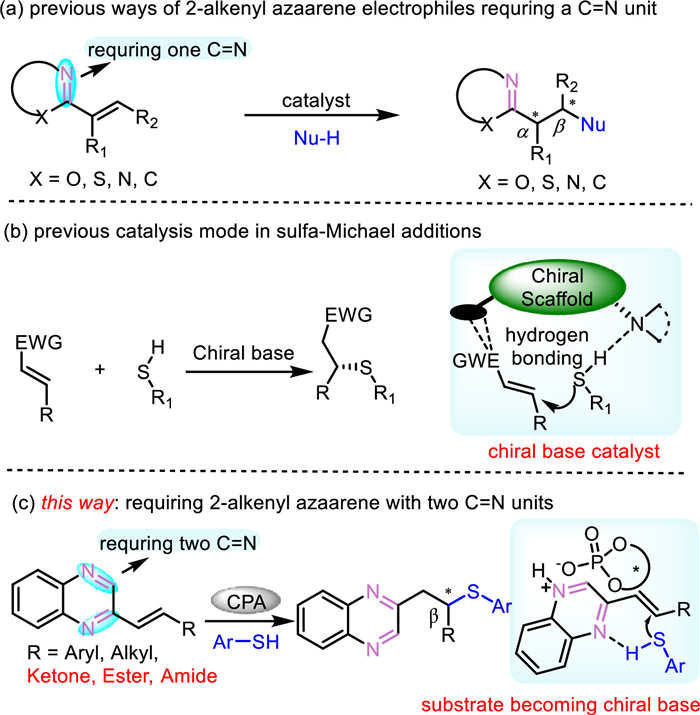
Scheme 1.
Proposal of the reaction.
Quinoxaline-specific enantioselective sulfa-michael reaction catalyzed by chiral phosphoric acid
Xiongfei Deng , Shiqi Zhang , Hesen Huang , Xin Cui , Zhuo Tang , Guangxun Li

Nitrogen-containing heteroarenes are privileged units in pharmaceutical molecules, advanced materials, chiral ligands, and natural products [1–9]. For example, quinoxalines are frequently incorporated in commercially available drugs such as chloroquinoxaline sulfonamide and brimonidine. Meanwhile, they are also contained in many biologically active compounds with antitumor activity, anti-glaucoma activity, and anti-inflammatory activity [10–15]. Therefore, significant efforts have been made in developing methods for the functionalization of azaarenes and their derivatives [16]. Among these useful methods, the direct exploitation of azaarenes bearing embedded C=N imine units as the analogs of weakened carbonyls to trigger the reaction, thus furnishing the enantioselective functionalization of prochiral azaarenes, has become an attractive method due to its potential advantages of using simple and readily accessible feedstocks and avoiding tedious operations in the late-stage modifications of products.
For instance, there are a variety of catalytic asymmetric methods using 2-alkylazaarenes as pronucleophiles via an enamine mechanism with respect to imine-containing azaarenes that feature an electron-withdrawing ability [17–20]. On the other hand, alkenyls adjacent to electron-deficient aromatic azaarenes have been extensively used as electrophiles in transition metal-catalyzed asymmetric C-C [21–24], C-B [25], C-Si bond formations [26,27], and organo-catalyzed asymmetric C-C [28], C-N [29,30], C-P [31], C-S bond formations [32]. In these elegant methods, the azaarene fragment plays an important role in activating the adjacent alkenyl to ensure the occurrence of the reactions. Moreover, one C=N unit in the aryl ring is associated with the generating of the enantioselectivity, while the other C=N unit seems needless to the reaction (Scheme 1a).
Asymmetric sulfa-Michael additions (SMA) play an important role in obtaining optically active thioethers, which are widely contained in pharmaceutics [33–35]. During the past decades, many elegant examples have been reported with electrophilic alkenyls adjacent to all sorts of electron-withdrawing groups including ketone [36–41], aldehyde [42–44], ester [45,46], amide [47,48] and nitro groups [49–52]. The bi-functional chiral base was proven to be the optimal catalyst for catalyzing these SMA reactions by deprotonating the ‒SH with the chiral base (Scheme 1b). On the other hand, Chiral phosphoric acid (CPA) has been widely used in activating alkenyl via LUMO-lowering protonation of the adjacent azaarenes. Therefore, N- and P-centered nucleophiles, and C-centered radicals have been applied in these enantioselective transformations, whereas the S-centered nucleophiles are seldom seen. To the best of our knowledge, there is only one enantioselective SMA reaction with alkenyl benzimidazoles as electrophiles and alkyl thiols as nucleophiles under bifunctional iminophosphorane catalysis [32]. Here, we found an interesting CPA-catalyzed SMA reaction, in which two C=N units are indispensable for controlling both the regioselectivity and enantioselectivity (Scheme 1c).
Our investigation begins with aromatic thiol 2a as a nucleophile and different alkenyl azaarenes 1 as electrophiles under the CPA catalyst (Scheme 2). According to the results, most of the alkenyl adjacent to different azaarenes such as quinoline, isoquinoline, benzeimidazole, and benzethiazoles could afford the corresponding products. However, negligible ee values were found in these products. In contrast, alkenyl adjacent to quinoxaline afforded product 7 in 86% yield and 80% ee.
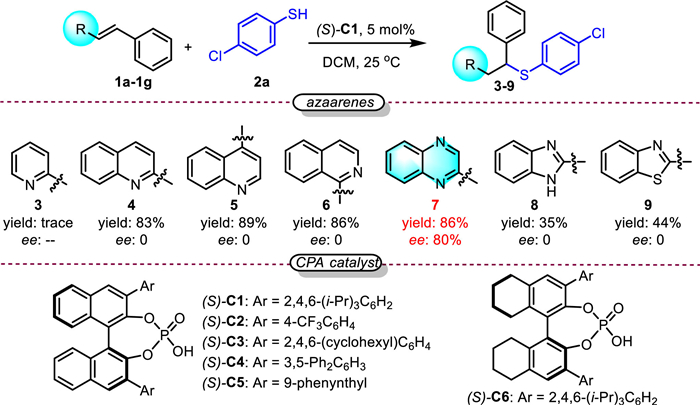
Based on this interesting discovery, we continued to optimize the reaction condition for obtaining chiral 7 (Table 1). Initially, a series of CPA catalysts were screened which proved that (S)-C6 was optimal for obtaining 7 in 91% yield and 82% ee (entries 1–6). Then the catalyst loading was investigated, which demonstrated that the reaction could catalyze with 1% catalyst with higher ee (entries 7 and 8). Next, the reaction solvent was screened and toluene was optimal for both increasing the yield and ee (entries 9 and 10). Finally, the ratio of the substrates, the concentration, temperature, additives, and reaction time were screened (Table S1 in Supporting information), which help us obtain the optimal reaction conditions: 0.1 mmol 1e, 0.2 mmol 2a, 1 mol% (S)-C6, 40 mg 3 Å molecular sieves as additives in 0.5 mL toluene at 30 ℃ under nitrogen for 8 h (entries 11–13). 7 could be obtained in 95% yield and 95% ee (entry 13).
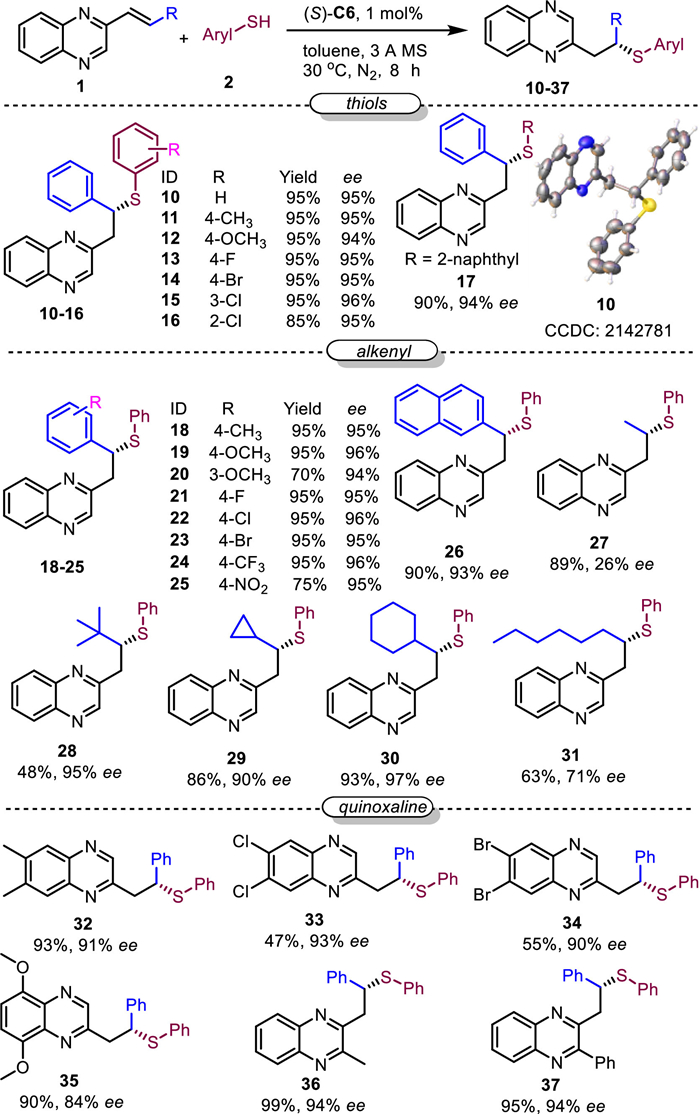
Under the optimal reaction condition, the scope of the reaction was investigated (Scheme 3). The scopes of aromatic thiols were broad including different substituents at p-, m-, and o-positions (10–16). Meanwhile, high yields (85%−95%) and excellent ee (94%−96%) were found with these substrates. Meanwhile, 2-naphthalenethiol with big steric hindrance was used as substrate and the corresponding product 17 was obtained in 90% yield and 94% ee. Next, the alkenyls were investigated, which demonstrated alkenyls adjacent to aryls with electron-donating groups (18–20), halogens (21–23), and electron-withdrawing groups (24 and 25) were compatible under the optimal condition. The corresponding products were obtained in good yields (70%−95%) and high ee (94%−96%). Meanwhile, alkenyl substituted with 2-naphthyl could afford the desired product 26 in 90% yield and 93% ee. Moreover, alkene with alkyl substituent was investigated, which revealed that small alkyl substituents such as methyl could afford the desired product 27 in 89% yield and 26% ee, while big substituents such as tert‑butyl afford the desired product 28 in 48% yield and 95% ee. Meanwhile, cyclic alkyls such as cyclopropyl and cyclohexyl, and long alkyl chains such as n-hexane, afforded the desired products 29–31 in good yields and ee values. Finally, the substituents of quinoxaline were investigated. The results demonstrated that substituents on the phenyl ring (32–35) and substituents on the hetero ring (36 and 37) are compatible with the reactions. The desired products were obtained in moderate to good yields (47%−99%) and good ee values (84%−94%).
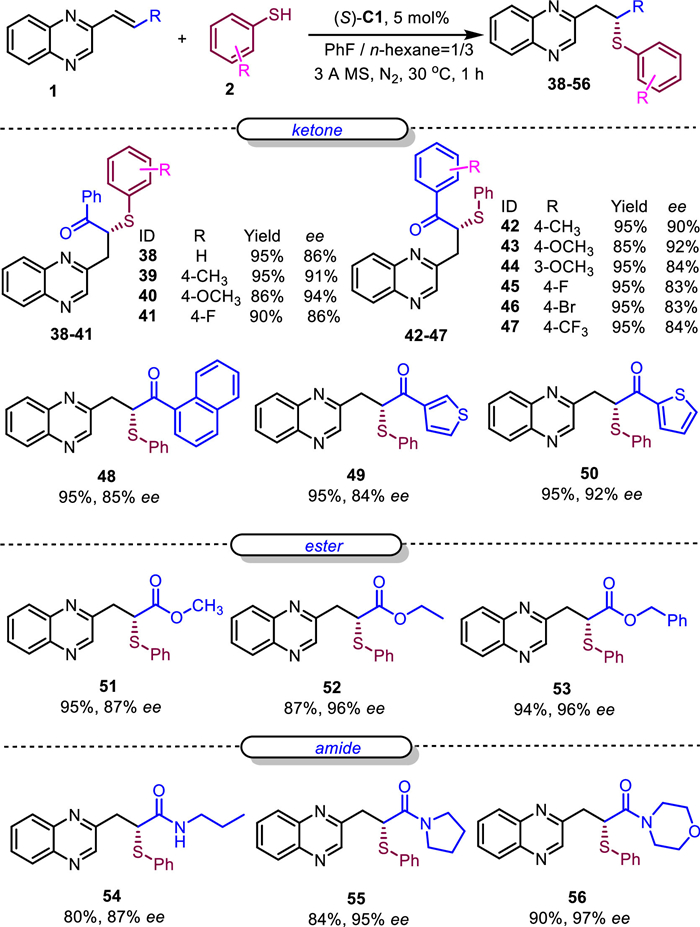
The above results indicated that quinoxaline was an efficient activation structure for LUMO lowering activating the adjacent alkenyl substituted with aryl or alkyl, for which the alkene was analogous to α, β-unsaturated ketones. Therefore, we wondered about the selectivity of the SMA reaction if the alkene was substituted with both quinoxaline and ketone. To our delight, the SMA reaction selectively occurred at the β position of quinoxaline and α position of carbonyl. This was both interesting and meaningful due to the fact that there were no efficient methods for obtaining the chiral α-tertiary sulfa ketones [53–57]. Therefore, we decided to optimize the reaction conditions (Table S2 in Supporting information). The desired product 38 could be obtained in 95% yield and 86% ee (Scheme 4). Then the reaction scopes of substrates were investigated and the results were displayed in Scheme 4. Firstly, the aromatic thiols were evaluated, which indicated that the phenyls with electron-donating groups afforded the desired products in higher ee (39 and 40). Then the substituents of aryl ketones were investigated. According to the results, phenyls with different substituents (42–47), sterically hindered α-naphthyl (48), thiophenyls (49 and 50), were compatible in delivering the corresponding products in good yields (85%−95%) and ee values (83%−92%). Then alkenes substituted with esters were evaluated, and the reaction selectively occurred at the β position of quinoxaline and α position of ester. The corresponding products 51–53 were obtained in good yields (87%−95%) and high ee (87%−96%). Finally, alkenes substituted with amides were investigated as well (54–56), and the reaction selectively occurred at the β position of quinoxaline and α position of amides. Meanwhile, good yields (80%−90%) and high ee values (87%−97%) were found in these substrates.
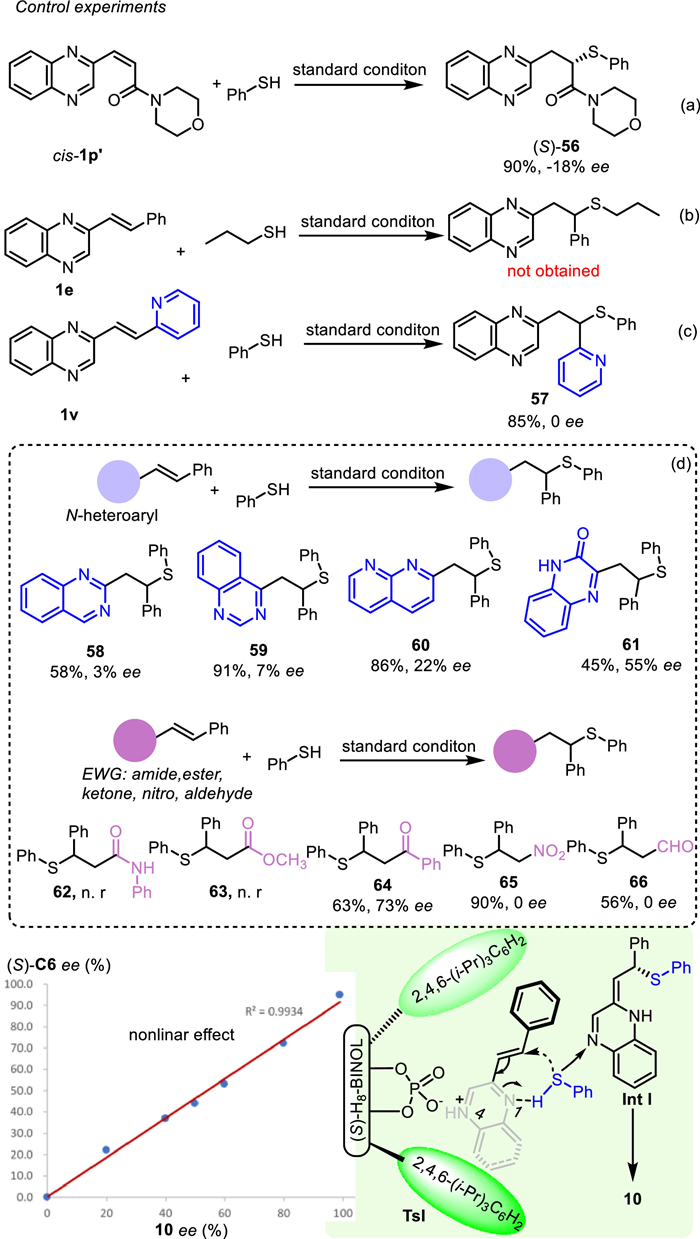
Then some preliminary control experiments were done so as to probe the plausible reaction mechanism (Scheme 5). Firstly, the cis-isomer of 1p' was investigated under the standard reaction condition, which revealed that the opposite isomer (S)−56 was obtained in 90% yield and 18% ee (Scheme 5a). This indicated that the reactivity of the olefin was not affected while the enantioselectivity was completely changed. Next, alkyl thiol was used as a substrate, while the desired product was not obtained which demonstrated that the C=N base was not sufficient to deprotonate the alkyl S-H (Scheme 5b). Meanwhile, alkene with quinoxaline and pyridine was used as substrate, and the desired product 57 was obtained as racemic, which might be ascribed to the formation of pyridine salts rather than quinoxaline salts because of the stronger alkalinity of pyridine (Scheme 5c). Next, a series of alkenes tethered with different azaarenes with two C=N units such as quinazoline, 1,8-naphthyridine, and 2-quinoxalinone, were investigated under the standard reaction conditions. The corresponding products (58–61) were obtained in moderate to high yields (45%−91%). Interestingly, low to moderate enantioselectivity was found in these substrates. This indicated that two C=N units were indispensable for controlling the reaction enantioselectivity. Meanwhile, the position of the C=N unit and the alkene was also important for the reaction stereoselectivity. Only the two C=N units located such as the quinoxaline could afford the product in higher enantioselectivity (61). On the hand, the electrophilic alkenyls adjacent to electron-withdrawing groups including ketone, aldehyde, ester, amide, and nitro groups were also studied under the standard conditions. We found the electrophilic alkenyls adjacent to amide 62 and ester 63 were not obtained under standard conditions. However, electrophilic alkenyls adjacent to aldehyde and nitro reacted under standard conditions and afforded the corresponding products 65 and 66 in pleasant yield and no ee. Interestingly, the electrophilic alkenyls adjacent to ketone reacted under standard conditions and afforded 64 with 63% yield and 73% ee. Furthermore, a linear relationship between the enantiopurity of the catalyst and product was found, which could suggest the involvement of one molecule of chiral phosphate in the enantioselective SMA reaction step. Based on the above results and previous work [30], we proposed a plausible reaction mechanism. Chiral ion pair (TS1) is formed from CPA and the N-4 of quinoxaline, and the aryl thiol is fastened by the N-1 of quinoxaline so as to form a six-ring transition state in the SMA reaction step. The aryl thiol adds to alkene from Si-face due to the steric hindrance of the 3,3′ substituent of C6 to afford the plausible intermediate Int I, which transforms to the desired product via re-aromatization.
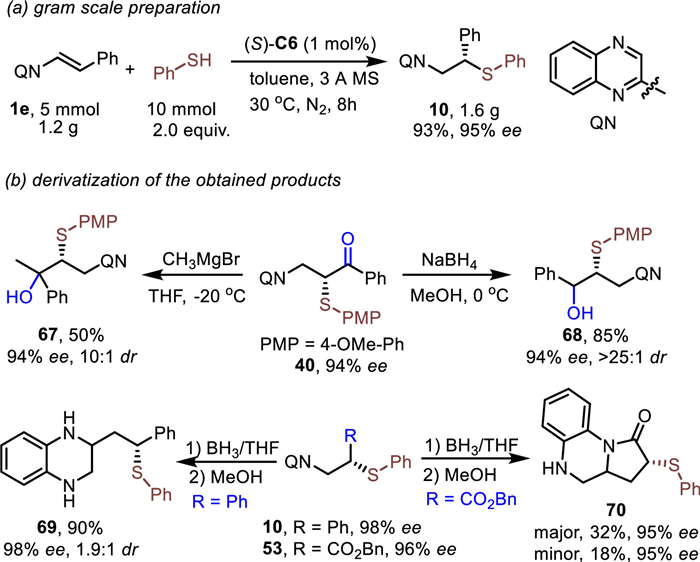
Next, we turned our attention to the application of the method. Firstly, the reaction could easily run on a gram scale for the preparation of the desired product in high efficiency with low catalyst loading (Scheme 6a). Then the transformations of the obtained products were investigated (Scheme 6b). The ketone could be converted to tertiary and secondary alcohol (67 and 68) via Grignard or a reducing reagent. The desired products were obtained in moderate to good yields (50%−85%), good ee (94%), and good dr (> 10:1). Meanwhile, the reduction of the quinoxaline ring was investigated. The desired product 69 could be obtained in 90% yield and 98% ee. To our delight, the lactam 70 could be obtained when 53 was used as substrate. The reaction may proceed via intramolecular amine ester exchange.
In summary, we have developed an interesting sulfa-Michael reaction that effectively proceeded under the low loading of the CPA catalyst. A series of azaarenes substituted non-terminal alkenes were investigated, which demonstrated that quinoxaline with two nitrogen atoms was a unique structure motif, for that only 2-alkenyl quinoxaline could afford the desired product in good enantioselectivity. Moreover, for non-terminal alkenes with both quinoxaline and other electro-withdrawing groups such as ketones, esters, or amides, the desired SMA reaction selectively occurred at the β-position of the quinoxaline. The reaction scope was very broad which allowed for the preparation of 48 new products in good yields (55%−99%) and ee (83%−97%). Meanwhile, the reaction could enlarge to a gram scale and allow for the transformation of other related compounds in high efficiency.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This project was supported by the West Light Foundation of the Chinese Academy of Sciences (No. 25E0C304) and the Sichuan Science and Technology Program (No. 2021ZYD0061).
Supplementary material associated with this article can be found, in the online version, at doi:
J. Yu, F. Shi, L.Z. Gong, Acc. Chem. Res. 44 (2011) 1156–1171. doi: 10.1021/ar2000343
X. Li, Q. Song, Chin. Chem. Lett. 29 (2018) 1181–1192. doi: 10.1016/j.cclet.2018.01.045
S. Li, S.H. Xiang, B. Tan, Chin. J. Chem. 38 (2020) 213–214. doi: 10.1002/cjoc.201900472
Y.C. Zhang, F. Jiang, F. Shi, Acc. Chem. Res. 53 (2020) 425–446. doi: 10.1021/acs.accounts.9b00549
B.C. Da, S.H. Xiang, B. Tan S. Li, Chin. J. Chem. 39 (2021) 1787–1796. doi: 10.1002/cjoc.202000751
H.H. Zhang, F. Shi, Acc. Chem. Res. 55 (2022) 2562–2580. doi: 10.1021/acs.accounts.2c00465
Z. Han, H. Zhuang, L. Tang, et al., Org. Lett. 24 (2022) 4246–4251. doi: 10.1021/acs.orglett.2c01559
F.T. Sheng, S. Yang, S.F. Wu, Y.C. Zhang, F. Shi, Chin. J. Chem. 40 (2022) 2151–2160. doi: 10.1002/cjoc.202200327
J.Y. Wang, S. Zhang, X.Y. Yu, et al., Tetrahedron Chem. 1 (2022) 100007. doi: 10.1016/j.tchem.2022.100007
Y.B. Kim, Y.H. Kim, J.Y. Park, S.K. Kim, Bioorg. Med. Chem. Lett. 14 (2004) 541–544. doi: 10.1016/j.bmcl.2003.09.086
A.S. Jaso, B.N. Zarranz, I. Aldana, A. Monge, J. Med. Chem. 48 (2005) 2019–2025. doi: 10.1021/jm049952w
X. Hui, J. Desrivot, C. Bories, et al., Bioorg. Med. Chem. Lett. 16 (2006) 815–820. doi: 10.1016/j.bmcl.2005.11.025
H.A. Abbas, A.R. Al-Marhabi, S.I. Eissa, Y.A. Ammar, Bioorg. Med. Chem. 23 (2015) 6560–6572. doi: 10.1016/j.bmc.2015.09.023
C.H. Tseng, Y.R. Chen, C.C. Tzeng, et al., Eur. J. Med. Chem. 108 (2016) 258–273. doi: 10.1016/j.ejmech.2015.11.031
L.A. Raphoko, K. Lekgau, C.M. Lebepe, et al., Bioorg. Med. Chem. Lett. 35 (2021) 127784. doi: 10.1016/j.bmcl.2021.127784
G. Yashwantrao, S. Saha, Org. Chem. Front. 8 (2021) 2820–2862. doi: 10.1039/D0QO01575J
B.M. Trost, D.A. Thaisrivongs, J. Am. Chem. Soc. 130 (2008) 14092–14093. doi: 10.1021/ja806781u
B.M. Trost, D.A. Thaisrivongs, J. Am. Chem. Soc. 131 (2009) 12056–12057. doi: 10.1021/ja904441a
B.M. Trost, D.A. Thaisrivongs, J. Hartwig, J. Am. Chem. Soc. 133 (2011) 12439–12441. doi: 10.1021/ja205523e
K.W. Bentley, K.A. Dummit, J.F. Van Humbeck, Chem. Sci. 9 (2018) 6440–6445. doi: 10.1039/C8SC00590G
G. Pattison, G. Piraux, H.W. Lam, J. Am. Chem. Soc. 132 (2010) 14373–14375. doi: 10.1021/ja106809p
I.D. Roy, A.R. Burns, G. Pattison, et al., Chem. Commun. 50 (2014) 2865–2868. doi: 10.1039/C4CC00340C
R.P. Jumde, F. Lanza, M.J. Veenstra, S.R. Harutyunyan, Science 352 (2016) 433–437. doi: 10.1126/science.aaf1983
R.P. Jumde, F. Lanza, T. Pellegrini, S.R. Harutyunyan, Nat. Commun. 8 (2017) 2058. doi: 10.1038/s41467-017-01966-7
L. Wen, Z. Yue, H. Zhang, Q. Chong, F. Meng, Org. Lett. 19 (2017) 6610–6613. doi: 10.1021/acs.orglett.7b03327
W. Mao, W. Xue, E. Irran, M. Oestreich, Angew. Chem. Int. Ed. 58 (2019) 10723–10726. doi: 10.1002/anie.201905934
Y.L. Zeng, B. Chen, Y.T. Wang, et al., Chem. Commun. 56 (2020) 1693–1696. doi: 10.1039/C9CC08910A
Y. Yin, Y. Dai, H. Jia, et al., J. Am. Chem. Soc. 140 (2018) 6083–6087. doi: 10.1021/jacs.8b01575
Y.Y. Wang, K. Kanomata, T. Korenaga, M. Terada, Angew. Chem. Int. Ed. 55 (2016) 927–931. doi: 10.1002/anie.201508231
C. Xu, C.W. Muir, A.G. Leach, A.R. Kennedy, A.J.B. Watson, Angew. Chem. Int. Ed. 57 (2018) 11374–11377. doi: 10.1002/anie.201806956
L. Hou, J. Kikuchi, H. Ye, M. Bao, M. Terada, J. Org. Chem. 85 (2020) 14802–14809. doi: 10.1021/acs.joc.0c01840
M. Formica, G. Sorin, A.J.M. Farley, et al., Chem. Sci. 9 (2018) 6969–6974. doi: 10.1039/C8SC01804A
D. Enders, K. Lüttgen, A. Narine, Synthesis 2007 (2007) 959–980. doi: 10.1055/s-2007-965968
P. Chauhan, S. Mahajan, D. Enders, Chem. Rev. 114 (2014) 8807–8864. doi: 10.1021/cr500235v
X. Liu, H. Wang, S. Li, et al., Chin. J. Org. Chem. 41 (2021) 3134–3143. doi: 10.6023/cjoc202103042
P. McDaid, Y. Chen, L. Deng, Angew. Chem. Int. Ed. 41 (2002) 338–340. doi: 10.1002/1521-3773(20020118)41:2<338::AID-ANIE338>3.0.CO;2-M
M. Moccia, F. Fini, M. Scagnetti, M.F. Adamo, Angew. Chem. Int. Ed. 50 (2011) 6893–6895. doi: 10.1002/anie.201102162
Y.P. Li, S.F. Zhu, Q.L. Zhou, Org. Lett. 21 (2019) 9391–9395. doi: 10.1021/acs.orglett.9b03615
X. Pan, Z. Wang, L. Kan, et al., Chem. Sci. 11 (2020) 2414–2419. doi: 10.1039/C9SC05894J
L. Wang, N. Wang, Y. Qi, et al., Chin. J. Org. Chem. 40 (2020) 3934–3943. doi: 10.6023/cjoc202004027
Z. Wang, Y. Zhu, X. Pan, G. Wang, L. Liu, Angew. Chem. Int. Ed. 59 (2020) 3053–3057. doi: 10.1002/anie.201912739
M. Marigo, T. Schulte, J. Franzeń, K.A. Jørgensen, J. Am. Chem. Soc. 127 (2005) 15710–15711. doi: 10.1021/ja055291w
S. Brandau, E. Maerten, K.A. Jørgensen, J. Am. Chem. Soc. 128 (2006) 14986–14991. doi: 10.1021/ja065507+
W. Wang, H. Li, J. Wang, L. Zu, J. Am. Chem. Soc. 128 (2006) 10354–10355. doi: 10.1021/ja063328m
D. Leow, S. Lin, S.K. Chittimalla, X. Fu, C.H. Tan, Angew. Chem. Int. Ed. 47 (2008) 5641–5645. doi: 10.1002/anie.200801378
X. Fang, J. Li, C.J. Wang, Org. Lett. 15 (2013) 3448–3451. doi: 10.1021/ol4015305
L. Zu, J. Wang, H. Li, et al., J. Am. Chem. Soc. 129 (2007) 1036–1037. doi: 10.1021/ja067781+
Y. Liu, B. Sun, B. Wang, M. Wakem, L. Deng, J. Am. Chem. Soc. 131 (2009) 418–419. doi: 10.1021/ja8085092
H.H. Lu, F.G. Zhang, X.G. Meng, S.W. Duan, W.J. Xiao, Org. Lett. 11 (2009) 3946–3949. doi: 10.1021/ol901572x
K.L. Kimmel, M.T. Robak, J.A. Ellman, J. Am. Chem. Soc. 131 (2009) 8754–8755. doi: 10.1021/ja903351a
X.F. Wang, Q.L. Hua, Y. Cheng, et al., Angew. Chem. Int. Ed. 49 (2010) 8379–8383. doi: 10.1002/anie.201004534
D. Uraguchi, N. Kinoshita, D. Nakashima, T. Ooi, Chem. Sci. 3 (2012) 3161– 3164. doi: 10.1039/c2sc20698f
S. Sobhani, D. Fielenbach, M. Marigo, T.C. Wabnitz, K.A. Jorgensen, Chem. Eur. J. 11 (2005) 5689–5694. doi: 10.1002/chem.200500512
L. Fang, A. Lin, H. Hu, C. Zhu, Chem. Eur. J. 15 (2009) 7039–7043. doi: 10.1002/chem.200901099
A. Lin, L. Fang, X. Zhu, C. Zhu, Y. Cheng, Adv. Synth. Catal. 353 (2011) 545–549. doi: 10.1002/adsc.201000679
Z. Han, W. Chen, S. Dong, et al., Org. Lett. 14 (2012) 4670–4673. doi: 10.1021/ol3021176
C. Wang, X. Yang, C.C. Loh, G. Raabe, D. Enders, Chem. Eur. J. 18 (2012) 11531–11535. doi: 10.1002/chem.201201262

Scheme 3 Investigation of the reaction scope with one electron-withdrawing group. Reaction conditions: 0.1 mmol 1, (S)-C6 (1 mol%) were dissolved in 0.5 mL toluene, then 0.2 mmol 2 and 40 mg 3 Å MS were added, and the resulting mixture was allowed to react under a nitrogen atmosphere at 30 ℃ for 8 h. Then the reaction was directly purified via silica gel columns and analyzed with HPLC with a chiral column.

Scheme 4 Investigation of the reaction scope with two electron-withdrawing groups. Reaction conditions: 0.1 mmol 1, (S)-C1 (5 mol%) were dissolved in 1 mL mix-solvent (fluorobenzene/n-hexane, 1:3), then 0.2 mmol 2 and 40 mg 3 Å MS were added, and the resulting mixture was allowed to react under a nitrogen atmosphere at 30 ℃ for 1 h. Then the reaction was directly purified via silica gel columns and analyzed with HPLC with a chiral column.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:


