
Figure 1.
Self-immolative linkers for phenol-containing compounds.
A mild phenoxysilyl linker for self-immolative release of antibody-drug conjugates
Ding Wei , Yurong Mao , Huihui Wang , Siqi Qu , Jiakang Chen , Jiusheng Li , Biao Jiang , Hongli Chen

Antibody-drug conjugates (ADCs) are emerging as one of the most promising targeted therapeutics [1-4]. By attaching a potent cytotoxic molecule to an antibody with an appropriate linker, ADCs are expected to specifically delivery the linked cytotoxin to the targeted cells with minimal toxicity. Although an increasing number of ADCs have demonstrated their efficiency and been approved by FDA, there are also many discontinued programs for ADCs due to the poor therapeutic windows. The chemical linker is one of the most important factors in ADCs development [5]. The linker is responsible for the release of cytotoxic molecules from ADCs and closely correlated with the efficiency and safety of ADCs [6,7]. Linkers are commonly classified as cleavable and non-cleavable. ADCs with non-cleavable linkers have no obvious chemical trigger to release the cytotoxic molecules and rely on the lysosomal proteolytic degradation to release the active molecule [8]. Whereas ADCs with cleavable linkers have clearly defined mechanisms to control the release of cytotoxic drugs [9]. Three main types of cleavable linkers have been well developed, including pH sensitivity, glutathione sensitivity and protease sensitivity linkers. The majority of current ADCs are preferred to use cleavable linkers because they have the ability to release the cytotoxic drugs through a controllable way. The development of other alternative linker with novel drug release mechanisms have been continually reported recently [10-19]. Phenol-containing compound is a kind of suitable agent to be masked and further triggered to be cleaved.
Several methods have been developed for the utilization of phenolic group to generate self-immolative unit, including carbonate, carbamate and ether linkages (Fig. 1). These linkage systems have been successfully applied to the activation of phenolic drugs [20,21] and some of them also have been employed as cleavable linkers to prepare ADCs [22,23]. In this study, we try to explore phenoxysilyl as a new self-immolative spacer to construct ADCs. Silyl ethers are widely used for the protection of alcohols. Recently, silyl ethers that linked to hydroxyl and amine containing molecules have been reported for ADCs development [24,25]. In this study, we further applied silyl ethers to directly modify the phenolic drug, combretastatin A4 (CA4). Atezolizumab, an FDA approved antibody that targets to the overexpression of PD-L1 was employed as targeting antibody. Then, phenoxysilyl based self-immolative ADC was prepared and its biological activities were evaluated. Phenoxysilyl linker is readily accessible and provides an alternative approach to the efficient release of precursors. More importantly, the release of the phenoxysilyl linker was fully investigated by mass spectrometry (MS), which would be helpful for the further design of silyl-containing linkers and also useful for the fuller understanding the effects of linkers to the related ADCs.
CA4, a microtubule depolymerizing agent with remarkable anticancer properties was chosen as the cytotoxic molecule, since it contained a phenolic group and we have employed it as the payload for the preparation of ADCs previously [26]. Using CA4 (1), dichlorodiisopropylsilane (2) and N3-PEG3-OH (3) as starting materials, a facile one-pot synthesis generated the desired phenoxysilyl linker 4. In previous studies, we have developed divinylsulfonamides (BVP to site-specifically construct ADCs through disulfide re-bridging approach [26-30]. Herein, BVP was also employed to react with compound 4 via click chemistry to generate the drug-linker PPS-CA4, which was further conjugated with atezolizumab to obtain the phenoxysilyl based ADC Ate-PPS-CA4. For comparison, a non-cleavable linker-based ADC was also prepared as the following procedure. CA4 was converted to its reactive carbonate 5, which was then condensed with tert-butyl(2, 5, 8, 11-tetraoxa-14-azahexadecan-16-yl)(methyl) carbamate to generate the intermediate 6. After deprotecting the Boc (tert-butoxycarbonyl) group, the resulting compound was reacted with N3-PEG3nullCOOH to afford compound 7. Similarly, after the reaction of click chemistry, the drug linker PBP-CA4 was obtained and further linked to the antibody, providing the ADC Ate-PBP-CA4 (Fig. 2a). We found that the silyl linker could improve the potential of CA4 might be due to its ability of the improvement of cell penetration (Fig. S5 in Supporting information). Also, the phenoxysilyl linker PPS-CA4 showed that it could increase the conjugation efficiency. Under the same reaction conditions, PPS-CA4 provided a higher DAR value. The data detected by mass spectrometry (MS) showed that DAR (drug-to-antibody ratio) 4 was the main product along with some DAR 2 and DAR 6 components for the ADC Ate-PPS-CA4. DAR 2 and DAR 4 products were obtained for the ADC Ate-PBP-CA4. The average DAR values of them were 3.45 and 2.96, respectively (Figs. 2b and c, Fig. S3 in Supporting information).

With the phenoxysilyl based ADC Ate-PPS-CA4 in hand, we began to carefully examine the release of the small molecule from it by MS. The ADC was incubated in phosphate buffer at 37 ℃ under two different pH values (pH 7.4 and 5.0). Aliquots were taken for LC-MS/MS analysis at the indicated time points. The drug was released with the prolonging of time. About 2 days later, ~50% of CA4 was released in pH 7.4 buffer (Fig. S1 in Supporting information). Whereas it was different from alkyl O-silyl ether, which was more liable to acidic conditions [25], the release of drug was slightly slow in pH 5 buffer for phenoxysilyl ether. This result was consistent with the fact that the release of silyl ether is strongly connected with electronics and phenoxysilyl linker is less susceptible to acidic conditions. What is more, the MS spectra clearly demonstrated how the small molecule released from the ADC. The cleavage started from the break of phenoxysilyl O-Si bond at a site that was prior to the cleavage of alkyl silyl O-Si bond at b site (Fig. 3a). Tiny levels of the released products were detected over 12 h. With the time going on, segments that linked to the silyl group were cleaved increasingly and more and more new peaks appeared (Fig. 3b).

Then the cytotoxic activities of the ADCs Ate-PPS-CA4 and Ate-PBP-CA4, along with the naked antibody atezolizumab and CA4 were evaluated against a panel of human cell lines expressing different surface densities of PD-L1. A fluorescence activated cell sorting (FACS)-based method was used to assess the levels of PD-L1 across cell lines (U87, Calu-1, MDA-MB-231, AsPC-1, MC38 and Raji). The result showed that the surface protein expression of PD-L1 was high in cells U87, Calu-1 and MDA-MB-231 and they were chosen to test the activities of the compounds mentioned above, along with the Raji and HepG2 as PD-L1 negative cell lines (Fig. S2 in Supporting information). The cell killing assays showed that Ate-PPS-CA4 exhibited good antiproliferative activities in PD-L1 positive cells (all the IC50 < 10 nmol/L, ) and it became less sensitive to Raji and HepG2 cells with IC50 = 25 nmol/L and 47 nmol/L respectively. Whereas the non-cleavable ADC Ate-PBP-CA4 exhibited low activity and the antibody displayed negligible inhibition effect against all the cell lines (Table 1).
 DownLoad:
CSV
DownLoad:
CSV

|
The ability for the self-immolative release and targeted drug delivery of Ate-PPS-CA4 was further investigated [31]. MDA-MB-231 and HepG2 as high and low PD-L1 expression cells were chosen and treated by Ate-PPS-CA4 (1 µmol/L), along with atezolizumab (1 µmol/L) and Ate-PBP-CA4 (1 µmol/L) for 30 min at 37 ℃. Then the cell media was removed. After the cells were washed with PBS, the new cell media was added and the cells were incubated for another 3 days at 37 ℃. This study was performed in triplicates. Ate-PPS-CA4 could effectively inhibit the growth of PD-L1 positive cells MDA-MB-231 and significant difference was observed compared with PD-L1 negative cells HepG2 (P < 0.01, Fig. 4a). While atezolizumab and Ate-PBP-CA4 exhibited negligible effect for the both cells. CA4 inhibited proliferative activities for both cells. This confirmed that Ate-PPS-CA4 could be targeted to PD-L1 protein and had the ability to self-immolative release of the active drug.
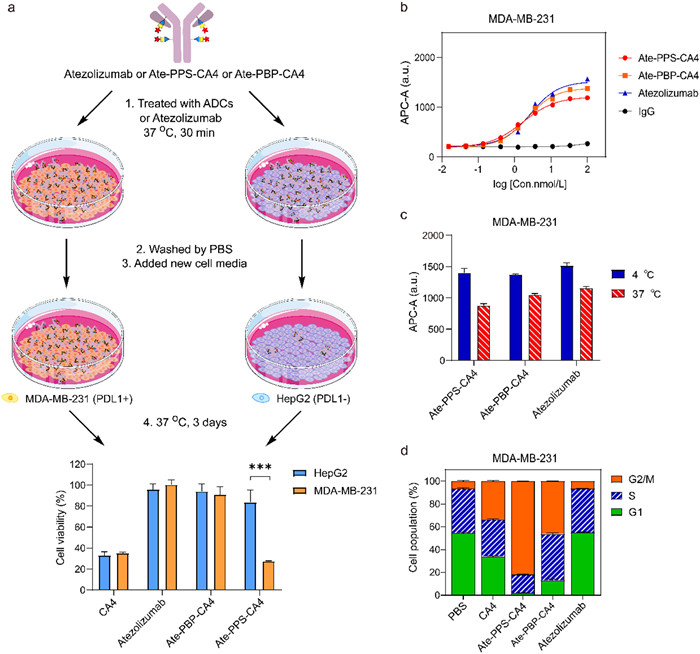
The binding affinity and internalization of ADCs Ate-PPS-CA4 and Ate-PBP-CA4 were tested by FACS on PD-L1 positive cells MDA-MB-231. Their binding affinity was similar with the naked antibody atezolizumab (Fig. 4b). Also, they retained the internalization feature that was comparable with atezolizumab (Fig. 4c). Whereas, negligible binding and internalization was observed when they treated PD-L1 negative cells HepG2 (Fig. S7 in Supporting information). It is well established that tubulin-destabilizing agents play an important role in the regulation of cell cycle progression [32,33]. FACS analysis was performed to determine the arrest effect of CA4 and ADCs. MDA-MB-231 cells were treated by the compounds at their IC50 concentration (Ate-PPS-CA4 at 8.5 nmol/L, Ate-PBP-CA4 at 900 nmol/L, CA4 at 2.5 nmol/L). The proportion of G2/M phase in CA4 and ADCs-treated cells was higher compared to vehicle PBS and atezolizumab, indicating that Ate-PPS-CA4 and Ate-PBP-CA4 arrest the cell cycle to the G2/M phase in keeping with CA4, a tubulin polymerization inhibitor (Fig. 4d). Microtubule imaging experiments were taken by a laser confocal microscope. Compared to vehicle PBS and atezolizumab for which the cells presented a typical elongated spindle shape (Lanes A and F in Fig. S9 in Supporting information), Ate-PPS-CA4, Ate-PBP-CA4 and CA4 induced changes in cell morphology at the dose of their IC50 (Lanes B-D in Fig. S9). When treated the cells by Ate-PBP-CA4 used the same dose of Ate-PPS-CA4 (Lane E in Fig. S9), the cells still retained the elongated spindle shape.
Linker is one of the most important elements for the construction of ADCs, which has significant impact on the biological activities, toxicity, stability and pharmacokinetics of ADCs. Development of linkers with different drug release mechanism is an attractive direction. In this study, a mild phenoxysilyl linker was developed for self-immolative release of phenolic-containing payload from ADCs. We demonstrated the utility of this phenoxysilyl linker to conjugate with CA4, a microtubule-disrupting drug, providing a new ADC Ate-PPS-CA4 with the ability to self-immolative release and targeted delivery. The process for the release of small molecules from Ate-PPS-CA4 was clearly detected by MS. It would be helpful for the design of next generation of silyl-containing prodrugs. The linker offers an alternative mechanism of release to expand ADC linker methods. It has the potential to be applied towards non-internalizing ADCs through extracellular cleavage. Since the release is highly relative to the electronics, we anticipated the rate of release can be tune by the choice of different functional groups. Further studies are necessary to explore the introduction of groups with different electronic moiety to control the rate of release.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This research was supported by Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine at ShanghaiTech University. We thank Analytical Chemistry platform (ShanghaiTech University, SIAIS) for the helpful technical assistance with LCMS and NMR experiments. We thank HPC Platform of ShanghaiTech University.
Z. Fu, S. Li, S. Han, C. Shi, Y. Zhang, Signal Transduct. Target. Ther. 7 (2022) 93. doi: 10.1038/s41392-022-00947-7
C.S.B. Chia, ChemMedChem. 17 (2022) e202200032.
S.J. Walsh, J.D. Bargh, F.M. Dannheim, A.R. Hanby, et al., Chem. Soc. Rev. 50 (2021) 1305–1353. doi: 10.1039/d0cs00310g
P. Tarantino, R.C. Pestana, C. Corti, et al., CA-Cancer J. Clin. 72 (2022) 165–182. doi: 10.3322/caac.21705
R. Sheyi, B.G. de la Torre, F. Albericio, Pharmaceutics 14 (2022) 396. doi: 10.3390/pharmaceutics14020396
M. Pabst, W. McDowell, A. Manin, et al., J. Control. Release 253 (2017) 160–164. doi: 10.1016/j.jconrel.2017.02.027
S. Cazzamalli, A.D. Corso, D. Neri, J. Control. Release 246 (2017) 39–45. doi: 10.1016/j.jconrel.2016.11.023
A. Dal Corso, S. Cazzamalli, R. Gebleux, M. Mattarella, D. Neri, Bioconjugate Chem. 28 (2017) 1826–1833. doi: 10.1021/acs.bioconjchem.7b00304
J.D. Bargh, A. Isidro-Llobet, J.S. Parker, D.R. Spring, Chem. Soc. Rev. 48 (2019) 4361–4374. doi: 10.1039/c8cs00676h
D.A. Rose, J.W. Treacy, Z.J. Yang, et al., J. Am. Chem. Soc. 144 (2022) 6050–6058. doi: 10.1021/jacs.2c01136
D. Kang, S. Lee, J. Kim, Chem. 8 (2022) 2260–2277. doi: 10.1016/j.chempr.2022.05.018
J. Li, D. Xiao, F. Xie, et al., Bioorg. Chem. 111 (2021) 104475. doi: 10.1016/j.bioorg.2020.104475
J.T. Miller, C.N. Vitro, S. Fang, S.R. Benjamin, L.N. Tumey, Bioconjugate Chem. 32 (2021) 842–858. doi: 10.1021/acs.bioconjchem.1c00124
J.P.M. Antonio, J.I. Carvalho, A.S. Andre, et al., Angew. Chem. Int. Ed. 60 (2021) 25914–25921. doi: 10.1002/anie.202109835
C. Zang, H. Wang, T. Li, et al., Chem. Sci. 10 (2019) 8973–8980. doi: 10.1039/c9sc03016f
R.V. Kolakowski, K.T. Haelsig, K.K. Emmerton, et al., Angew. Chem. Int. Ed. 55 (2016) 7948–7951. doi: 10.1002/anie.201601506
X.B. Ma, S.J. Li, Y.T. Liu, et al., Chem. Soc. Rev. 51 (2022) 5136–5174. doi: 10.1039/d2cs00247g
S. Bai, L.L. Yang, Y.J. Wang, et al., Small 16 (2020) 2000214. doi: 10.1002/smll.202000214
T. Liu, H. Zou, J.Q. Mu, et al., Chin. Chem. Lett. 32 (2021) 1751–1754. doi: 10.1016/j.cclet.2020.12.008
B.E. Toki, C.G. Cerveny, A.F. Wahl, P.D. Senter, J. Org. Chem. 67 (2002) 1866–1872. doi: 10.1021/jo016187+
D. Shabat, H.N. Lode, U. Pertl, et al., Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 7528–7533. doi: 10.1073/pnas.131187998
R.C. Elgersma, R.G. Coumans, T. Huijbregts, et al., Mol. Pharm. 12 (2015) 1813–1835. doi: 10.1021/mp500781a
S.V. Govindan, T.M. Cardillo, R.M. Sharkey, et al., Mol. Cancer. Ther. 12 (2013) 968–978. doi: 10.1158/1535-7163.MCT-12-1170
Y. Wang, S. Fan, D. Xiao, et al., Cancers 11 (2019) 957. doi: 10.3390/cancers11070957
M.C. Finniss, K.S. Chu, C.J. Bowerman, et al., Med. Chem. Commun. 5 (2014) 1355–1358. doi: 10.1039/C4MD00150H
R. Huang, Y. Sheng, Z. Xu, et al., Eur. J. Med. Chem. 216 (2021) 113355. doi: 10.1016/j.ejmech.2021.113355
D. Wei, Y. Mao, Z. Xu, et al., Bioorgan. Med. Chem. 51 (2021)116497. doi: 10.1016/j.bmc.2021.116497
R. Huang, Y. Sheng, D. Wei, et al., Eur. J. Med. Chem. 190 (2020) 112080. doi: 10.1016/j.ejmech.2020.112080
R. Huang, Y. Sheng, D. Wei, et al., Bioorgan. Med. Chem. 28 (2020) 115793. doi: 10.1016/j.bmc.2020.115793
Z. Li, R. Huang, H. Xu, J. Chen, et al., Org. Lett. 19 (2017) 4972–4975. doi: 10.1021/acs.orglett.7b02464
A. Marcher, M.A.D. Nijenhuis, K.V. Gothelf, Angew. Chem. Int. Ed. 60 (2021) 21691–21696. doi: 10.1002/anie.202107221
Y. Yang, X.N. Zhu, Y.B. Chen, X.L. Wang, R.Z. Chen, Eur. J. Pharmacol. 576 (2007) 26–33. doi: 10.1016/j.ejphar.2007.07.067
C. Schmidt, T. Babu, H. Kostrhunova, et al., J. Med. Chem. 64 (2021) 11364–11378. doi: 10.1021/acs.jmedchem.1c00706

Figure 2 Preparation of ADCs Ate-PPS-CA4 and Ate-PBP-CA4. (a) The scheme for the synthesis of drug-linkers PPS-CA4 and PBP-CA4. Reagents and conditions: (i) imidazole, 4-dimethylaminopyridine, N, N-dimethylformamide (DMF), room temperature (r.t.), 1.5 h; (ii) 4-nitrophenyl chloroformate, pyridine, dichloromethane (DCM), 0 ℃, 4 h; (iii) tert-butyl (2, 5, 8, 11-tetraoxa-14-azahexadecan-16-yl)(methyl) carbamate, trimethylamine (TEA), 1, 2-dichloroethane, 40 ℃, 24 h; (iv) trifluoroacetic acid (TFA), DCM, r.t., 4 h; 2-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)acetic acid, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 1-hydroxybenzotriazole (HOBt), 4-methylmorpholine (NMM), r.t., overnight; (v) BVP, l-ascorbic acid sodium, CuSO4, tBuOH/H2O/DMF (1:1:1), r.t., 1 h. (b) Synthesis of Ate-PPS-CA4 and Ate-PBP-CA4. (c) MS analysis for Ate-PPS-CA4 and Ate-PBP-CA4.

Figure 3 The release of small molecules from ADC Ate-PPS-CA4. (a) The sites for cleavage. (b) The process of the fragmentation detected by MS.

Figure 4 Activities evaluation for Ate-PPS-CA4. (a) Targeted drug delivery: MDA-MB-231 (PD-L1 positive cell line) and HepG2 (PD-L1 negative cell line) were treated with Ate-PPS-CA4 (1 µmol/L) or Ate-PBP-CA4 (1 µmol/L) or atezolizumab (1 µmol/L) and washed with PBS and finally cell viability was tested. ***P < 0.01, cell viability of HepG2 vs. MDA-MB-231; Value = Mean ± SEM, n = 3. (b) Binding affinity of Ate-PPS-CA4, Ate-PBP-CA4 and atezolizumab in MDA-MB-231 cell. (c) Internalization of Ate-PPS-CA4, Ate-PBP-CA4 and atezolizumab in MDA-MB-231 cell. (d) The effects of Ate-PPS-CA4, Ate-PBP-CA4, atezolizumab and CA4 on cell cycle arrest.
Table 1. Cytotoxicity of ADCs, atezolizumab and CA4 against different cell lines in vitro.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
