Scheme 1.
Palladium-catalyzed C—H bond acetylation.
Phosphine oxide directing-group-enabled atroposelective C–H bond acyloxylation via an eight-membered palladacycle intermediate
Peng-Bo Bai , Ming-Ying Wu , Xin-Xin Yang , Gang-Wei Wang , Shang-Dong Yang

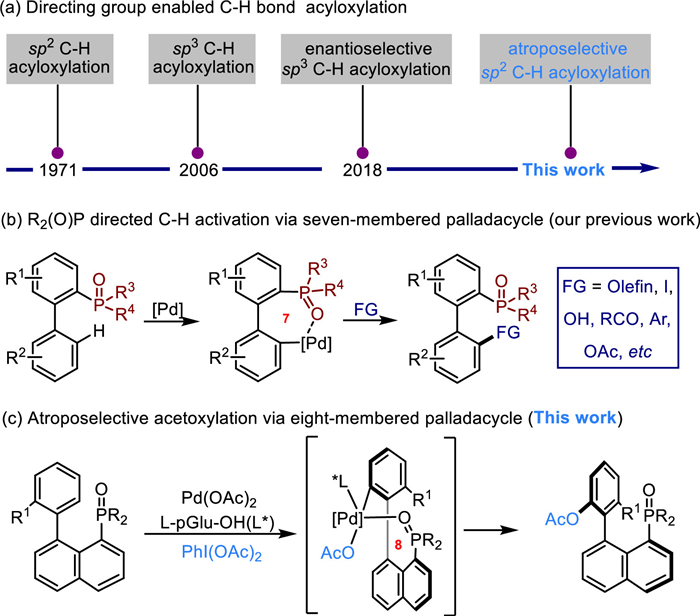
Transition-metal-catalyzed direct C–H bond functionalizations that are enabled by a suitable directing group, permit a step and atom-economical approach for accessing complex molecular targets in a regioselective manner [1-14]. Within this area, C(sp2)-H bond acyloxylation is among the earliest research topics, with Henry reporting their seminal discovery in 1971 [15-17]. Such transformations are highly useful, since acetyl groups can be selectively introduced and can serve as useful functional handles for further diversification, allowing facile access to decorated aromatic compounds. The innovation and modification of such transformations has therefore attracted great interest since then, and huge successes have been achieved. For example, both Yu and Corey groups independently reported C(sp3)-H bond acyloxylations in 2006, and the Yu group also completed an elegant enantioselective C–H bond acyloxylation in 2018 (Scheme 1a) [18-20]. In addition, the Sahoo, Sanford, and Dong groups have also made significant contributions to the area [21-25]. To our surprise, to date there are scarcely any reports related to atroposelective C(sp2)-H bond acyloxylation [26], especially given the fact that atroposelective C(sp2)-H bond functionalization has become a powerful tool to access axially chiral biaryl skeletons that found broad application in natural products, advanced materials, as well as in synthetic chemistry as privileged chiral ligands and catalysts [27-41].
The value of directing-group-enabled transition-metal-catalyzed C–H functionalization methodologies is improved when the directing-groups itself owns devise important functionalities [42,43]. Among others, organophosphorus compounds have broad applicationsin agrochemistry, clinical drugs, and biological sciences [44-46]. As a result, the study of their construction and transformations is significantly import for the synthetic community [47-49]. In 2013, our group initiated a program that utilizes phosphine oxide and phosphinate directing group to realize C(sp2)-H bond functionalizations using palladium catalysis. This method included C–H olefination, iodination, hydroxylation, acylation, and arylation, as well as acetoxylation (Scheme 1b). A variety of important phosphorus-containing compounds, including useful biphenyl-phosphine ligands that contain axial chirality and/or a chirogenic phosphorus center, were produced efficiently [50-59].
Uniquely, this transformation is shown to proceed via a kinetically and thermodynamically disfavored seven-memberedpalladacycle intermediate, thus providing a complementary pathway to the commonly proposed, more stable, and more easily formed five- or six-membered metallacycle intermediates enabled by C—H bond functionalization [60-64]. To further broadenthe utility of the above phosphine oxide and phosphinate directed C–H bond functionalization, we considered its extension to an eight-membered palladacycle mediated transformation. The success of such a process would not only lead to a mechanistically distinguished C–H bond functionalization, but also provide facile access to a variety of important organophosphorus compounds that would otherwise be difficult to prepare by current methodologies, for example, the preparation of novel axially chiral compounds through such a medium ring sized palladacycle intermediate enabled remote C—H bond functionalization. However, this proposal is recognizably challenging, as the formation of an eight-membered palladacycle is even slower and energetically demanding, as a result of the more significant transannular interactions andentropic factors [65,66]. In addition, the difficulties with achieving asymmetric induction through the use of chiral ligands with these less stable metallacycles are also increased [67-77]. Herein, we have developed the first phosphine-oxide-directed atroposelective C(sp2)-H bond acyloxylation reaction, which is achieved via an uncommon eight-membered palladacycle intermediate (Scheme 1c). The cheap and abundant chiral aminoacid derivative L-pGlu-OH was identified as the optimal chiral ligand, delivering generally excellent enantioselective control. The products of this method can easily be further transformed into a diverse range of potential chiral ligands.
To test the feasibility of our proposed eight-membered palladacycle enabled atroposelective C(sp2)-H bond acyloxylation, we began our studies by using diphenyl(8-(o-tolyl)naphthalen-1-yl)phosphine oxide (1a) as the model substrate in a reaction with commercially available (diacetoxyiodo)benzene, which serves as both an oxidant and a source of acetate (Table 1). After an extensive screening of metal catalysts, chiral ligands, reaction solvents, and reaction time, the optimal conditions we obtained were: Pd(OAc)2 (5 mol%), L1 (40 mol%) in TFE and HFIP (5:1, 0.025 mol/L), at 60 ℃ for 18 h under air (entry 1), which afforded the desired product 2a in an 82% yield with 95% enantiomeric excess (ee). The absolute R configuration of 2a was determined by an X-ray crystallo-graphic analysis (CCDC: 2130450). Apart from L-pGlu-OH, most other amino acid ligands that were tested proved to be ineffective in controlling the reaction enantioselectivity (entries 2–6). The transformation was completely shut down when Pyox or Box type ligands were used (entries 7 and 8). Increasing the catalyst loading from 5 mol% to 10 mol% resulted in a slightly higher final yield, but with a diminished ee value (entry 9). The loading of the chiral ligand L-pGlu-OH (L1) was critical, and the ee value obtained fell dramatically when lower amounts of L1 were used (entries 10 and 11). Finally, the reaction solvent-system appeared to have little effect on the outcome of the reaction (entries 12–14).
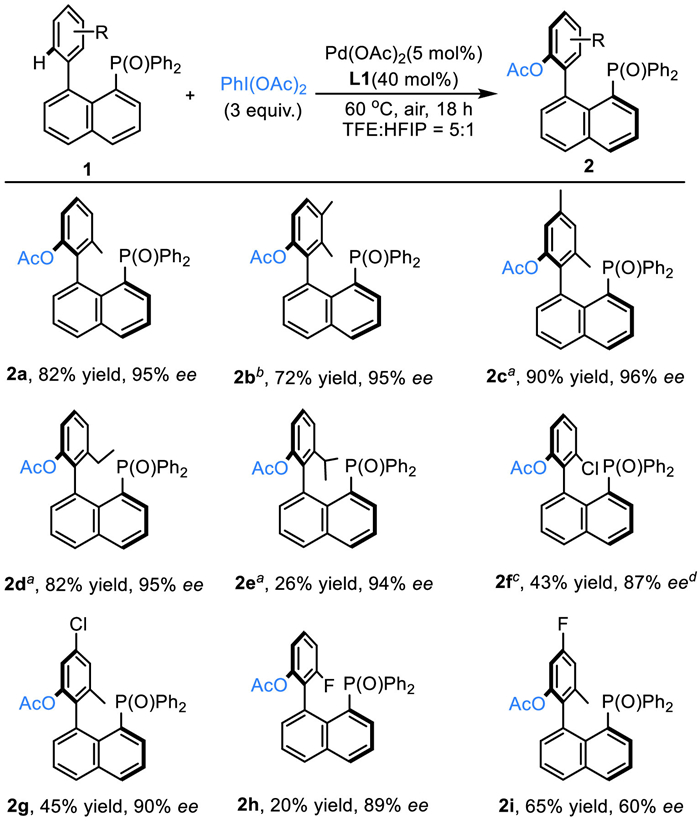
With the optimal reaction conditions in hand, the scope of substitution on the binaphthene unit was investigated (Scheme 2). Substrates bearing one or two electron-donating groups were smoothly transformed into the desired atroposelective C—H acyloxylation products with good yield and excellent ee values (2a-2d). We also found that increased steric bulk of the substituent had a large detrimental impact on the yield of the product, while the enantioselectivity was not affected at all (2e). Consistent with the observation we made in our previous phosphine oxide directed C–H functionalization procedure, the current method also delivered the products in lower yields when electron-withdrawing substituents were introduced, however, relatively good enantioselective control was still achieved (2f-2i). In addition, the endeavor to extent the scope to heteroaromatic-based substrates was failed.
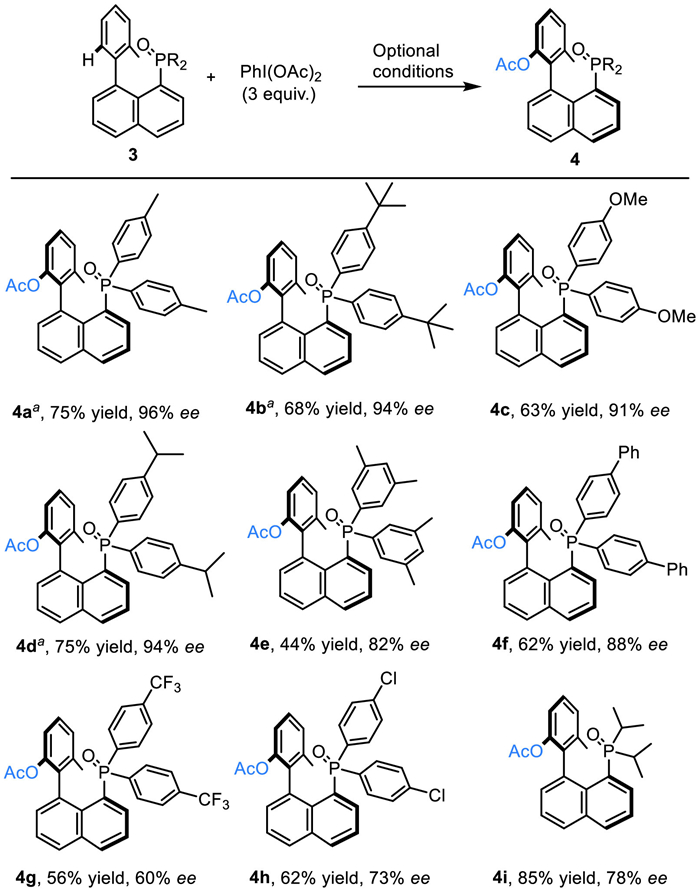
Next, we turned our attention to substrates bearing different P-substituents (Scheme 3). This substitution is considered important as it allows the preparation of diverse phosphorus-containing compounds which bear an axial chirality. Indeed, phosphine oxides with methyl, tert–butyl, methoxy or isopropyl substituents on the phenyl ring were all able to function as the directing group, and the desired products were obtained in moderate yields with good to excellent enantioselective control (4a-4d). As observed previously, increasing steric bulk led to a negative effect on the outcome of the reaction, with 3,5-dimethyl-substituted diphenylphosphine oxide giving product 4e in a reduced 44% yield, with 82% ee. To our delight, diphenylphosphine oxides bearing electron withdrawing groups were also suitable directing groups for this reaction, demonstrating a clear tendency towardslower yields and ee values with electron withdrawing substituents (4f-4h). Notably, in addition to diphenylphosphine oxide derivatives, alkyl-substituted phosphineoxides were also able to be utilized as directing groups capable of performing the atroposelective C–H acyloxylation reaction in high yields, albeit with relatively lower enantioselective control (4i).
To evaluate the practicality of our strategy, a gram-scale experiment was conducted, delivering 2a with a slightly reduced yield, but without any erosion of enantioselectivity (60%, 95% ee) (Scheme 4). One of the key features of our directed aromatic C–H acyloxylation reactions is that they allow the selective introduction of an acetyl group, which can then serve as a functional handle for further diversification. Indeed, product 2a could be easily converted into axially chiral biaryl 5, which bears a hydroxyl group, under mild conditions, with excellent yield and full preservation of enantiopurity. Further transformations of 5 were also conducted to showcase its synthetic potential. Again, good to excellent yields and high enantiopurities for triflation, methylation, and reduction of phosphine oxide 5 were achieved, generating products 6, 7 and 8. These possess axially chiral phosphorus-oxygen backbones, and hence can potentially be used as chiral ligands.

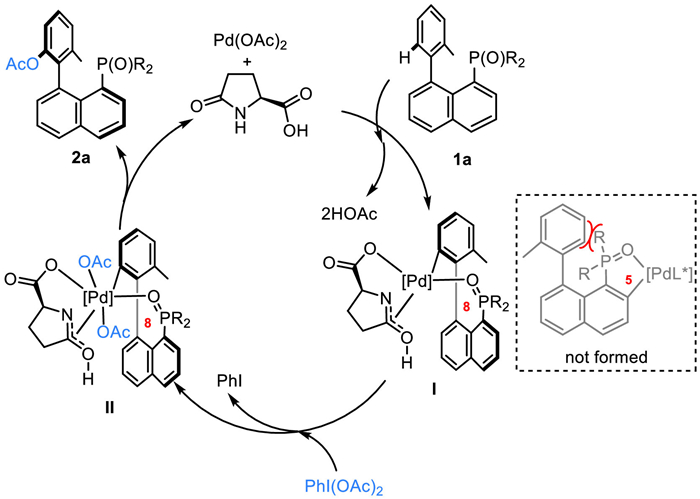
In accordance with our previous work and other related literature [78,79], a possible mechanism for the current process is proposed (Scheme 5). Initially, with the assistance of L-pGlu-OH, the phosphine oxide of 1a directs the C–H palladation with Pd(OAc)2 to form a chiral eight-membered palladacycle intermediate I, presumably going via a concerted-metalation-deprotonation (CMD) C—H bond activation process. Next, oxidative addition of PhI(OAc)2 to Ⅰ occurs, generating Pd(IV) intermediate Ⅱ. After that, reductive elimination of Ⅱ takes place to afford the desired chiral phosphate-acetyl compound 2a, as well as closing the catalytic cycle. It is worth noting that phosphine oxide in 1a could also direct transition metaltogo through a C—H palladation with its ortho-C-H bond, to form a kinetically favored five-membered metallacycle intermediate, thus leading to an ortho-C–H bond acyloxylation reaction [60-64]. However, such a process was completely subdued in our system, and we did not observe any of the associated products. We attributed this outcome to the thermodynamically instability of the five-membered metallacycle intermediate which is resulted from steric repulsion between substitution on phosphine oxide and the phenyl group (Scheme 5).
In summary, we have developed the first atroposelective sp2 C—H bond acyloxylation reaction. The transformation was enabled by a phosphine oxide directing-group, and likely proceeds through a unique eight-membered palladacycle intermediate, as opposed to a kinetically and thermodynamically favored five-membered palladacycle intermediate. Moreover, commercially available and inexpensive chiral amino acid derivative L-pGlu-OH was used as the chiral ligand, and this process yielded a variety of axially chiral phosphonoacetyl compounds, which can potentially be used as novel chiral ligands in asymmetric reactions.
Authors of Peng-Bo Bai, Ming-Ying Wu, Xin-XinYang, Gang-Wei Wang, and Shang-DongYang declare that there are no conflicts in the paper of Phosphine Oxide Directing-Group-Enabled Atroposelective C–H Bond Acyloxylationvia an Eight-membered PalladacycleIntermediate.
We are grateful to the Natioal Natural Science Foundation of China (NSFC, No. 22171119), Gansu Province Science and Technology Plan Project (Nos. 21YF5WA114 and 21ZD4WA021) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
K.M. Engle, T.S. Mei, M. Wasa, J.Q. Yu, Acc. Chem. Res. 45 (2012) 788–802. doi: 10.1021/ar200185g
C.J. Li, Acc. Chem. Res. 42 (2009) 335–344. doi: 10.1021/ar800164n
C.J. Scheuermann, Chem. Asian J. 5 (2010) 436–451. doi: 10.1002/asia.200900487
L. Ackermann, Chem. Rev. 111 (2011) 1315–1345. doi: 10.1021/cr100412j
B. Liu, A.M. Romine, C.Z. Rubel, K.M. Engle, B.F. Shi, Chem. Rev. 121 (2021) 14957–15074. doi: 10.1021/acs.chemrev.1c00519
X.L. Han, P.P. Lin, Q.J. Li, Chin. Chem. Lett. 30 (2019) 1495–1502. doi: 10.1016/j.cclet.2019.04.027
N. Kuhl, M.N. Hopkinson, J. Wencel-Delord, F. Glorius, Angew. Chem. Int. Ed. 51 (2012) 10236–10254. doi: 10.1002/anie.201203269
B.J. Li, Z.J. Shi, Chem. Soc. Rev. 41 (2012) 5588–5598. doi: 10.1039/c2cs35096c
S.A. Girard, T. Knauber, C.J. Li, Angew. Chem. Int. Ed. 53 (2013) 74–100.
J. Wencel-Delord, F. Glorius, Nat. Chem. 5 (2013) 369–375. doi: 10.1038/nchem.1607
T.W. Lyons, M.S. Sanford, Chem. Rev. 110 (2010) 1147–1169. doi: 10.1021/cr900184e
L.M. Xu, B.J. Li, Z. Yang, Z.J. Shi, Chem. Soc. Rev. 39 (2010) 712–733. doi: 10.1039/B809912J
Y. Xu, G.B. Dong, Chem. Sci. 9 (2018) 1424–1432. doi: 10.1039/c7sc04768a
M. Zhang, S.L. Zhong, Y.Y. Peng, et al., Org. Chem. Front. 8 (2021) 133–168. doi: 10.1039/d0qo00988a
P.M. Henry, J. Org. Chem. 36 (1971) 1886–1890. doi: 10.1021/jo00813a009
T. Yoneyama, R.H. Crabtree, J. Mol. Catal. A 108 (1996) 35–40. doi: 10.1016/1381-1169(95)00289-8
K. Muñiz, Angew. Chem. Int. Ed. 48 (2009) 9412–9423. doi: 10.1002/anie.200903671
D.H. Wang, X.S. Hao, D.F. Wu, J.Q. Yu, Org. Lett. 8 (2006) 3387–3390. doi: 10.1021/ol061384m
B.V.S. Reddy, L.R. Reddy, E.J. Corey, Org. Lett. 8 (2006) 3391–3394. doi: 10.1021/ol061389j
H. Park, P. Verma, K. Hong, J.Q. Yu, Nat. Chem. 10 (2018) 755–762. doi: 10.1038/s41557-018-0048-1
R.K. Rit, M.R. Yadav, A.K. Sahoo, Org. Lett. 14 (2012) 3724–3727. doi: 10.1021/ol301579q
K.J. Stowers, A. Kubota, M.S. Sanford, Chem. Sci. 3 (2012) 3192–3195. doi: 10.1039/c2sc20800h
A.K. Cook, M.H. Emmert, M.S. Sanford, Org. Lett. 15 (2013) 5428–5431. doi: 10.1021/ol4024248
Z. Ren, F.Y. Mo, G.B. Dong, J. Am. Chem. Soc. 134 (2012) 16991–16994. doi: 10.1021/ja3082186
Y. Xu, G.B. Yan, Z. Ren, G.B. Dong, Nat. Chem. 7 (2015) 829–834. doi: 10.1038/nchem.2326
J. Zhang, J. Fan, Y.H. Wu, et al., Org. Lett. 24 (2022) 5143–5148. doi: 10.1021/acs.orglett.2c01981
J.K. Cheng, S.H. Xiang, S. Li, L. Ye, B. Tan, Chem. Rev. 121 (2021) 4805–4902. doi: 10.1021/acs.chemrev.0c01306
Y.J. Wu, G. Liao, B.F. Shi, Green. Syn. Catal. 3 (2022) 117–136. doi: 10.1016/j.gresc.2021.12.005
G. Liao, T. Zhou, Q.J. Yao, B.F. Shi, Chem. Commun. 55 (2019) 8514–8523. doi: 10.1039/c9cc03967h
G.J. Mei, W.L. Koay, C.Y. Guan, Y. Lu, Chem 8 (2022) 1855–1893. doi: 10.1016/j.chempr.2022.04.011
X. Zhang, K. Zhao, Z. Gu, Acc. Chem. Res. 55 (2022) 1620–1633. doi: 10.1021/acs.accounts.2c00175
S. Fang, J.P. Tan, J. Pan, et al., Angew. Chem. Int. Ed. 60 (2021) 14921–14930. doi: 10.1002/anie.202102352
C. Nájera, F. Foubelo, J.M. Sansano, M. Yus, Org. Biomol. Chem. 18 (2020) 1279–1336. doi: 10.1039/c9ob02597a
J.J. Petkowski, W. Bains, S. Seager, Molecules 24 (2019) 866–931. doi: 10.3390/molecules24050866
Y. Dong, R. Liu, W. Wang, Green. Syn. Catal. 1 (2020) 83–85. doi: 10.1016/j.gresc.2020.09.002
X.D. Qiu, M.Y. Wang, Y. Zhao, Z.Z. Shi, Angew. Chem. Int. Ed. 56 (2017) 7233–7237. doi: 10.1002/anie.201703354
W.Q. Ji, H.H. Wu, J.L. Zhang, ACS Catal. 10 (2020) 1548–1554. doi: 10.1021/acscatal.9b04354
X. Zhang, J. Wang, S.D. Yang, ACS Catal. 11 (2021) 14008–14015. doi: 10.1021/acscatal.1c04128
X.H. Wei, C.Y. Bai, L.B. Zhao, et al., Chin. J. Chem. 39 (2021) 1855–1860. doi: 10.1002/cjoc.202100083
Q. Feng, X.X. Ma, W. Bao, et al., CCS Chem. 3 (2021) 377–387. doi: 10.31635/ccschem.021.202000725
Q. Zhang, L.S. Wu, B.F. Shi, Chem 8 (2022) 384–413. doi: 10.1016/j.chempr.2021.11.015
W.P. Liu, L. Ackermann, ACS Catal. 6 (2016) 3743–3752. doi: 10.1021/acscatal.6b00993
J. He, M. Wasa, K.S.L. Chan, et al., Chem. Rev. 117 (2017) 8754–8786. doi: 10.1021/acs.chemrev.6b00622
A. Hosseinian, S. Farshbaf, L.Z. Fekri, M. Nikpassand, E. Vessally, Top. Curr. Chem. 376 (2018) 23–41. doi: 10.1007/s41061-018-0200-9
S. Demkowicz, J. Rachon, M. Daśko, W. Kozak, RSC Adv. 6 (2016) 7101–7112. doi: 10.1039/C5RA25446A
N. Sbei, G.M. Martins, B. Shirinfar, N. Ahmed, Chem. Rec. 20 (2020) 1530–1552. doi: 10.1002/tcr.202000096
R. Morodo, P. Bianchi, J.C.M. Monbaliu, Eur. J. Org. Chem. 33 (2020) 5236–5277. doi: 10.1002/ejoc.202000430
V.B. Silva, Y.H. Santos, R. Hellinger, et al., Green Chem. 24 (2022) 585–613. doi: 10.1039/d1gc02705k
M.A. Shameem, A. Orthaber, Chem. Eur. J. 22 (2016) 10718–10735. doi: 10.1002/chem.201600005
Y.N. Ma, S.X. Li, S.D. Yang, Acc. Chem. Res. 50 (2017) 1480–1492. doi: 10.1021/acs.accounts.7b00167
H.L. Wang, R.B. Hu, H. Zhang, et al., Org. Lett. 15 (2013) 5302–5305. doi: 10.1021/ol402577p
H.Y. Zhang, H.M. Yi, G.W. Wang, et al., Org. Lett. 15 (2013) 6186–6189. doi: 10.1021/ol403028a
H. Zhang, R.B. Hu, X.Y. Zhang, S.X. Li, S.D. Yang, Chem. Commun. 50 (2014) 4686–4689. doi: 10.1039/C4CC01238K
Z.C. Qi, Q.X. Lou, Y. Niu, S.D. Yang, Chem. Commun. 57 (2021) 2021–2024. doi: 10.1039/d0cc07596e
R.B. Hu, H. Zhang, X.Y. Zhang, S.D. Yang, Chem. Commun. 50 (2014) 2193–2195. doi: 10.1039/C3CC49050E
Y.N. Ma, Q.P. Tian, H.Y. Zhang, A.X. Zhou, S.D. Yang, Org. Chem. Front. 1 (2014) 284–288. doi: 10.1039/C4QO00005F
S.X. Li, Y.N. Ma, S.D. Yang, Org. Lett. 19 (2017) 1842–1845. doi: 10.1021/acs.orglett.7b00608
J. Wang, P.B. Bai, S.D. Yang, Chin. Chem. Lett. 33 (2022) 2397–2401. doi: 10.1016/j.cclet.2021.10.019
Y. Niu, C.X. Yan, X.X. Yang, et al., Org. Chem. Front. 9 (2022) 1023–1032. doi: 10.1039/d1qo01454d
S. Takebayashi, T. Shibata, Organometallics 31 (2012) 4114–4117. doi: 10.1021/om300348e
M. Itoh, Y. Hashimoto, K. Hirano, et al., J. Org. Chem. 78 (2013) 8098–8104. doi: 10.1021/jo401393b
C.S. Wang, P.H. Dixneuf, J.F. Soulé, ChemCatChem 9 (2017) 3117–3120. doi: 10.1002/cctc.201700557
Z. Liu, J.Q. Wu, S.D. Yang, Org. Lett. 19 (2017) 5434–5437. doi: 10.1021/acs.orglett.7b02710
Q.X. Lou, Y. Niu, Z.C. Qi, et al., J. Org. Chem. 85 (2020) 14527–14536. doi: 10.1021/acs.joc.0c00999
G. Illuminati, L. Mandolini, Acc. Chem. Res. 14 (1981) 95–102. doi: 10.1021/ar00064a001
N.L. Allinger, M.T. Tribble, M.A. Miller, D.H. Wertz, J. Am. Chem. Soc. 93 (1971) 1637–1648. doi: 10.1021/ja00736a012
W.F. Maier, P.V.R. Schleye, J. Am. Chem. Soc. 103 (1981) 1891–1900. doi: 10.1021/ja00398a003
Y.J. Hu, L.X. Li, J.C. Han, et al., Chem. Rev. 120 (2020) 5910–5953. doi: 10.1021/acs.chemrev.0c00045
R.L. Reyes, T. Iwai, M. Sawamura, Chem. Rev. 121 (2021) 8926–8947. doi: 10.1021/acs.chemrev.0c00793
E.J. Corey, K.C. Nicolaou, J. Am. Chem. Soc. 96 (1974) 5614–5616. doi: 10.1021/ja00824a073
B. Biletskyi, P. Colonna, K. Masson, et al., Chem. Soc. Rev. 50 (2021) 7513–7538. doi: 10.1039/d0cs01396j
S.J. Miller, S.H. Kim, S.J. Miller, et al., J. Am. Chem. Soc. 117 (1995) 2108–2109. doi: 10.1021/ja00112a031
B. Zhou, Y.Q. Zhang, K. Zhang, et al., Nat. Commun. 10 (2019) 3234–3244. doi: 10.1038/s41467-019-11245-2
A.K. Clarke, W.P. Unsworth, Chem. Sci. 11 (2020) 2876–2881. doi: 10.1039/d0sc00568a
L. Huang, L.X. Dai, S.L. You, J. Am. Chem. Soc. 138 (2016) 5793–5796. doi: 10.1021/jacs.6b02678
M.J. Ralph, D.C. Harrowven, S. Gaulier, S. Ng, K.I. Booker-Milburn, Angew. Chem. Int. Ed. 54 (2015) 1527–1531. doi: 10.1002/anie.201410115
Q.J. Yao, P.P. Xie, Y.J. Wu, et al., J. Am. Chem. Soc. 142 (2020) 18266–18276. doi: 10.1021/jacs.0c09400
L. Jin, Q.J. Yao, P.P. Xie, et al., Chem 6 (2020) 497–511. doi: 10.1016/j.chempr.2019.12.011
Y.J. Wu, P.P. Xie, G. Zhou, et al., Chem. Sci. 12 (2021) 9391–9397. doi: 10.1039/d1sc01130h

Scheme 2 Substrate scope of the aromatic ring of binaphthene bearing P(O)Ph2 directing group. Reaction conditions: 1 (0.1 mmol, 1.0 equiv.), PhI(OAc)2 (0.3 mmol, 3 equiv.), Pd(OAc)2 (0.005 mmol, 5 mol%), and L1 (0.04 mmol, 40 mol%) in TFE and HFIP (5:1, 4 mL, 0.025 mol/L) at 60 ℃ for 18 h under air; Isolated yield. The ee value was determined by chiral HPLC. a The reaction was conducted at 50 ℃. b The reaction was conducted at 40 ℃. c Pd(OAc)2 (0.01 mmol, 10 mol%), and L1 (0.06 mmol, 60 mol%).

Scheme 3 Substrate scope of the P-sources. Reaction conditions: 3 (0.1 mmol, 1.0 equiv.), PhI(OAc)2 (0.3 mmol, 3 equiv.), Pd(OAc)2 (0.005 mmol, 5 mol%), and L1 (0.04 mmol, 40 mol%) in TFE and HFIP (5:1, 4 mL, 0.025 mol/L) at 60 ℃ for 18 h under air; isolated yield. The ee value was determined by chiral HPLC. a The reaction was conducted at 50 ℃.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

