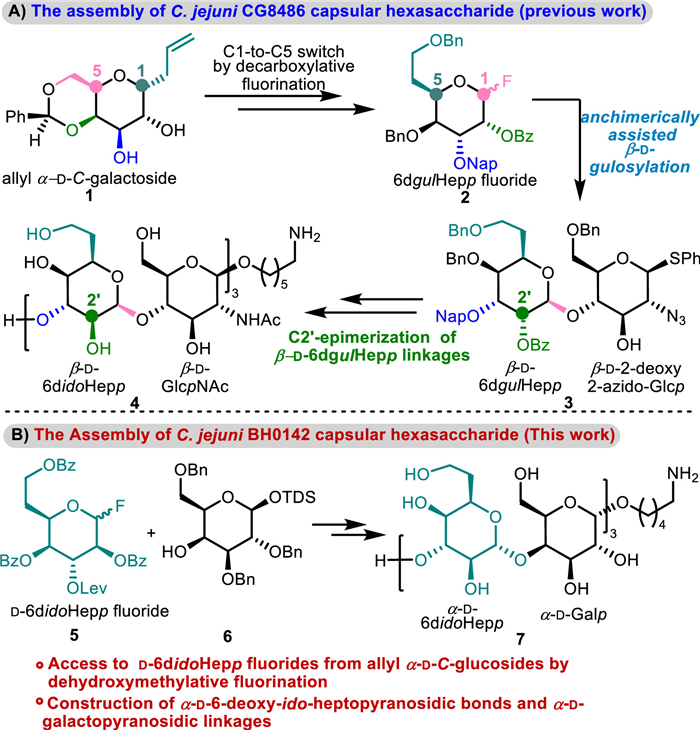
Scheme 1.
Synthesis of C. jejuni strain CG8486 and BH0142 capsular hexasaccharides.
Synthesis of a conjugable hexasaccharide corresponding to the capsular polysaccharide of Campylobacter jejuni strain BH0142
Zijiao Hou , Jianjun Wang , Xinxin Zhang , Peng Wang , Ni Song , Ming Li

Campylobacter jejuni (C. jejuni) is one of the leading causes of human gastroenteritis worldwide [1]. Although campylobacteriosis is both preventable and treatable in most cases, it still poses enormous challenges to animal and public health. C. jejuni infections can result in serious sequelae such as a rare autoimmune disease known as Guillain–Barré Syndrome [2] and reactive arthritis [3]. It is estimated that C. jejuni is linked to 40% of all new cases of Guillain–Barré Syndrome [4]. Campylobacter infections are associated with the death of around 530,000 children every year [5]. Globally, resistance to clinical antibiotics against C. jejuni infections is increasingly rising and therefore creates the urgent need for alternative therapeutics.
Glycoconjugate vaccines derived from native and synthetic glycans have been recognized as efficient tools to combat pathogenic infections and to stem antibiotic resistance crisis [6,7]. A preeminent example in point is the approved semi-synthetic vaccine against Haemophilus influenzae Type B [8]. The C. jejuni capsular polysaccharides (CPSs) are the key antigenic determinants of the Penner serotyping scheme [9] and represent potential vaccine targets [10,11]. Natural C. jejuni CPS-based glycoconjugate vaccines showed significant immunogenicity in animal test [12], highlighting the potential of such conjugates in discovery and development of new therapeutics to combat C. jejuni infections. These advantageous properties might be linked to unique structures of C. jejuni CPSs, which are commonly embedded by uncommon d/l-glycero-d/l-heptosyl units or related 6-deoxy derivatives [13]. Among them, the glycans containing 6-deoxy-d-ido-heptopyranosyl (6didoHepp) units and its l-glycero congeners caught our attention because these constructs have not been found in other organisms.
Considerable efforts have been devoted to development of glycoconjugate vaccines for the prevention of campylobacteriosis in the past 20 years, however, there are no approved vaccines available [10,14]. We recently established a novel C1-to-C5 switch strategy for the synthesis of uncommon d/l-6-deoxy-heptopyranosyl fluorides from the easily accessible allyl α-C-hexopyranosides relying on oxidative radical decarboxylative fluorination of uronic acids [15]. The transformation has been applied in the first assembly of a hexasaccharide related to the C. jejuni. strain CG8486 capsular polysaccharide (Scheme 1A) consisting of →3)-β-d-6didoHepp-(1→4)-β-d-GlcpNAc-(1→. The synthesis features the preparation of 6dgulHepp fluoride from allyl α-d-C-galactoside and the indirect construction of the challenging 1,2-cis-β-d-idopyranosides by epimerizing the C2 configuration of 1,2-trans-β-d-gulopyranosides [15,16].
Given that there are 35 types of C. jejuni CPSs recognized so far, it is desirable to make multivalent glycoconjugate vaccines based on different glycans to prevent pathogenesis of C. jejuni [17]. As part of our efforts to culminate in synthesis and biological evaluation of structurally defined multivalent synthetic vaccines against C. jejuni infections, we herein describe blockwise synthesis of a conjugable hexasaccharide 7 corresponding to the C. jejuni. strain BH0142 capsular polysaccharide composed of →3)-α-d-6didoHepp-(1→4)-α-d-Galp-(1→ (Scheme 1B). This assembly is characterized by the convenient preparation of unique d-6didoHepp fluoride from the readily available allyl α-d-C-glucoside through oxidative radical dehydroxymethylative fluorination, the convenient construction of 1,2-trans-α-d-ido-heptopyranosidic bonds relying on the catalytic activation of orthogonally protected idopyranosyl fluoride, and of the stereoselective formation of 1,2-cis-α-d-galactopyranosidic linkages.
Structurally, target molecule 7 is a trimer derivative of disaccharide repeating unit. Accordingly, we chose to take a convergent approach using a {2 + [2 + 2]} strategy to reach the goal. As shown in Scheme 2, we attempted to generate 7 by unmasking the fully protected hexasaccharide 8. The assembly of 8 could be achieved by iterative employment of the key disaccharide N-phenyl trifluoroacetimidate (PTFA) 9 as glycosylating agent through stereoselective formation of 1,2-cis-α-d-galactopyranosidic linkages with aglycone 10 [18] and the C3-OH of idosyl moiety. Disaccharide donor 9 could be traced back to 11 bearing a temporary protecting group dimethylthexylsilyl (TDS) at the reducing end. The preparation of 11 entailed stereocontrolled construction of 1,2-trans-α-d-glycosidic bonds between 6didoHepp donor with galactosyl acceptor.
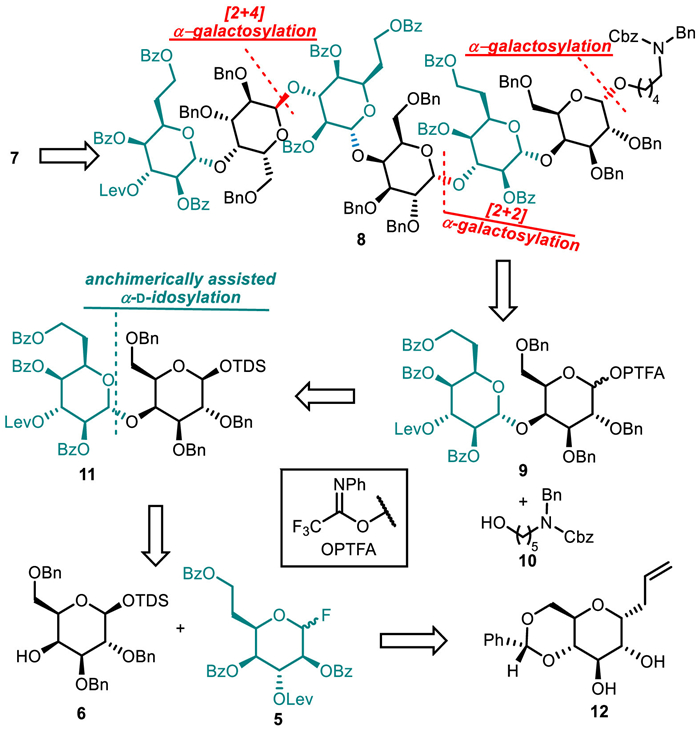
Inspired by the work of Pakulski who successfully applied benzoyl (Bz)-protected idopyranosyl donor in synthesis of α-d-idosides [19], we envisaged that the desired disaccharide 11 could be achieved by means of the anchimerically assisted glycosylation with 6-deoxy-d-ido-heptopyranosyl donor bearing C2-, C4- and C7-benzoates. In addition, in order to extend the sugar chain at C3 position of idosyl moiety, an orthogonal and temporary protecting group should be installed at that position. On the other hand, the construction of α-d-galactopyranosidic linkage required the incorporation of C2-ether type substituent. Furthermore, to increase the nucleophilicity of the axial C4-OH of Galp units favoring glycosylation at that sterically hindered position, the masking of the hydroxy groups adjacent to that group with ether-type protecting group should be preferred. With those considerations in mind and inspired by synthesis of rare glycosyl fluorides by use of radical dehydroxymethylative fluorination [20,21] as well as their catalytic activation [22] to engage in glycosylation, we would like to use 6didoHepp fluoride 5 as glycosyl donor to introduce the 6didoHepp residue and the literature-known compound 6 [23] as the galactosyl building block that is protected by benzyl (Bn) groups at C2-, C3- and C6-OHs. Fluoride 5 could be prepared proceeding from allyl α-C-glucopyranoside 12 [15] by a C1-to-C5 switch strategy through oxidative radical dehydroxymethylative fluorination as a key transformation.
Our synthesis commenced with the preparation of d-6didoHepp fluoride 5 (Scheme 3). We initially attempted to use 9-fluorenylmethyloxy carbonyl (Fmoc) to mask the C3-OH of 6didoHepp moiety because of the reaction conditions for its installation and deprotection leaving benzoates intact, and of its tolerance of both Zn/AcOH- and NaBH4-mediated reduction reaction (vide infra). To this end, diol 12 was subjected to intramolecular iodoetherification [24], the ensuing reaction with FmocCl, and Zn/AcOH-promoted reductive ring opening [25] of iodomethyl tetrahydrofuran. The reaction sequence delivered the desired product 13 in 83% yield over three steps with the liberation of the C2-OH and the recovery of the anomeric allyl substituent. At this stage, benzoylation of 13 was uneventfully achieved with BzCl in the presence of 0.01 equiv. of 4-N, N-dimethylpyridine (DMAP) in pyridine. The reaction afforded the expected benzoate 14 in 94% yield with Fmoc group intact. It is important to note that the larger amounts of DMAP caused the removal of the Fmoc group, producing thus 2,3-di-O-benzoate S2 (see Supporting information).
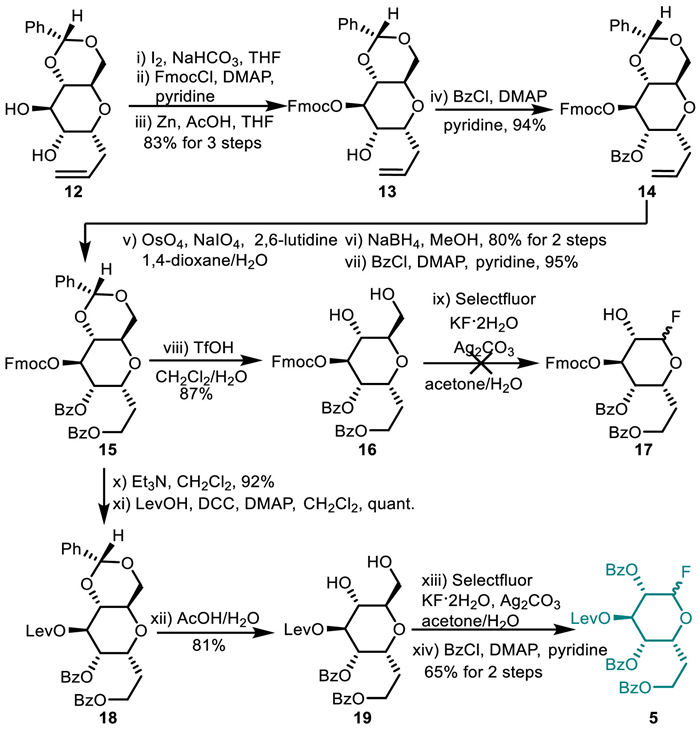
Oxidative cleavage of the olefin using OsO4/NaIO4 in aqueous dioxane, subsequent reduction of the resulting aldehyde using NaBH4, and benzoylation converted 14 into benzoate 15 in 76% yield. The removal of the benzylidene in 15 using trifluoromethanesulfonic acid (TfOH)-catalyzed transacetalization in MeOH afforded diol 16 in 87% yield. Exposure of 16 to Ag2CO3-induced radical dehydroxymethylative fluorination reaction [20,21], however, resulted in a mess reaction. We assumed that this unrewarding result might be attributed to lability of Fmoc group to the used conditions. To address this issue, we resorted to levulinoyl (Lev) to protect the C3-OH of 6didoHepp unit because of its tolerance of radical dehydroxymethylative fluorination reaction. Levulinate 18 was achieved by the cleavage of Fmoc with Et3N in CH2Cl2 and the subsequent introduction of Lev using dicyclohexylcarbodiimide (DCC)-mediated condensation with levulinic acid [26]. Chemoselective radical dehydroxymethylative fluorination of the primary hydroxy group over the secondary one in diol 19, prepared by deprotecting the benzylidene in 18, afforded the desired glycosyl fluoride as an anomeric mixture of α/β 2/1. At this point, the required d-6didoHepp fluoride 5 was prepared in 29% overall yield involving a 12-step reaction sequence from allyl α-d-C-glucoside 12.
With fluoride 5 in hand, we next focused on glycosylation of 6 [23] with 5 to generate the requisite disaccharide imidate 9 (Scheme 4). We recently found that 0.1 equiv. of (C6F5)3B·(HF)n as catalyst enable glycosylation of various alcohol acceptors with disarmed glycosyl fluorides [22]. To our delight, the (C6F5)3B·(HF)n-catalyzed coupling reaction of 5 with 6 smoothly and stereoselectively afforded the desired disaccharide 11 in 71% yield due to neighboring group participation of the C2-benzoate in 5. The anomeric 1JC-H 174 Hz of the idosyl unit is diagnostic of 1,2-trans-α-d-configuration of the newly formed glycosidic bond. In addition, the silyl group TDS was found to survive the glycosylation conditions. Upon treatment with acetic acid-buffered tetrabutylammonium fluoride (TBAF) followed by reaction of the resulting hemiacetal with N-phenyltrifluoroacetiminoyl chloride 21 in the presence of K2CO3 in acetone, compound 11 was transformed into glycosyl N-phenyltrifluoroacetimidate donor 9 in 71% isolated yield over two steps.

With disaccharide donor 9 secured, we moved our attention to the equipment of the capping linkage at the reducing end by constructing 1,2-cis-α-d-galactopyranosidic linkages, the formation of which is not trivial, especially for relatively reactive primary alcohols. Various tactics are adopted to address this issue including employment of the anomeric effects [27], judicious choice of the protecting groups [28], recourse to the solvent effects [29]. As such, we evaluated the effects of solvents, catalysts, and reaction temperatures on the outcome of the coupling between 9 and 10. As compiled in Table S1 (Supporting information), the results revealed that trimethylsilyl perchlorate (TMSClO4)-catalyzed glycosylation of 9 with 10 in ether furnished glycosides 22 and 22 in the highest 5.6/1 stereoselectivity and 91% total yield, favoring the α-isomer as the expected product. We attributed these observations to the cooperation of the solvent effects of ether and the utility of perchlorate counterions of the catalyst. Both of them have proven to favor the formation of the axial-type glycosidic bonds [30,31].
With disaccharides 9 and 22 in hand, we set out to conduct the sugar chain assembly. As depicted in Scheme 5, the orthogonal cleavage of the levulinate using hydrazine acetate in 22 proceeded well to provide 23 in an excellent yield of 91%, ready for glycosylation at its C3′-OH. Trimethylsilyl trifluoromethanesulfonate (TMSOTf)-catalyzed coupling of 23 with 9 in ether gave rise to tetrasaccharide 24 in 61% yield with exclusive α-stereoselectivity. Compared to glycosylation of 10, this improved stereochemical outcome might be associated with the weaker nucleophilicity of the axially-oriented secondary C3′-OH in 23 than the primary hydroxy group of 10, which shift the glycosylation reaction toward a SN1-like process [32]. The same manipulations as those of 24 involving the cleavage of levulinate and glycosylation with 9, tetrasaccharide 25 was converted into hexasaccharide 8 in 86% yield.
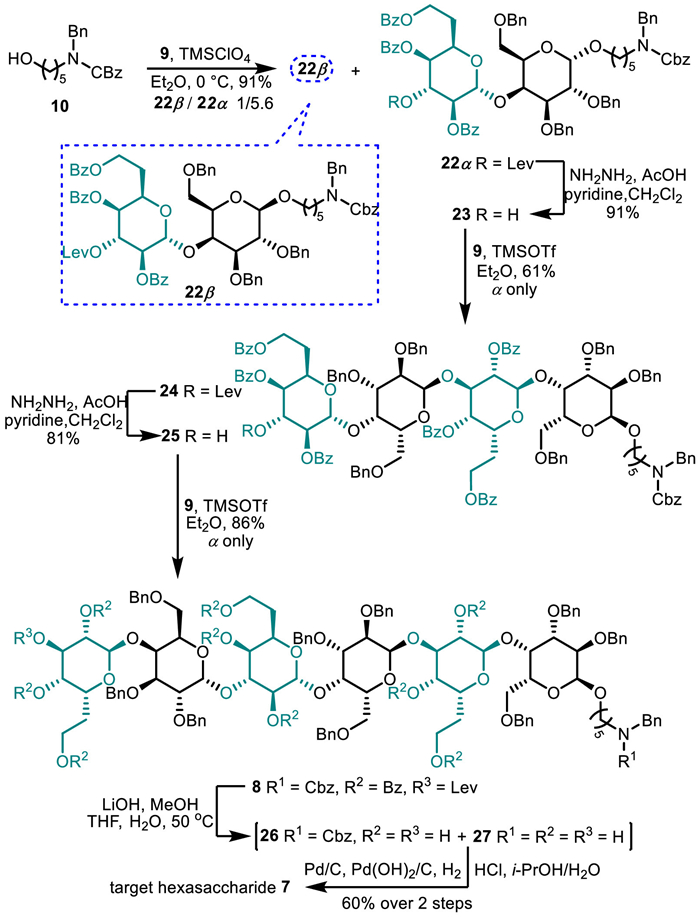
Next, deprotection of 8 en route to target glycan 7 was attempted. For this end, we exposed 8 to LiOH-promoted hydrolysis of esters in THF/H2O at 50 ℃. After stirring overnight, the mass spectrometric analysis of the crude reaction solution showed that the reaction resulted in a mixture of 26 with complete cleavage of the benzoates and the levulinate as well as 27 with additional deprotection of the benzyloxy carbamate (Cbz). Due to difficulty in separating 27 from 26 by silica gel column chromatography, the mixture was treated with Pd(OH)2/C and Pd/C under hydrogen atmosphere in i-PrOH/H2O at room temperature. The hydrogenolysis reaction proceeded smoothly and provided target molecule 7 in 60% yield over two steps. The structure of 7 was corroborated by the extensive 1D and 2D NMR experiments (Supporting information), and the configuration of the constructed glycosidic bonds was verified by the corresponding anomeric 1JC-H coupling constants (for α-d-6didoHepp bonds, 1JC1-H1 = 167.6 and 168.2 (overlapped) Hz; for α-d-Galp bonds, 1JC1-H1 = 171.2 (overlapped) and 171.3 Hz).
In conclusion, we have accomplished the first assembly of a hexasaccharide corresponding to the capsular polysaccharide of C. jejuni strain BH0142. The synthesis features the expeditious preparation of the orthogonally protected 6didoHepp fluoride and (C6F5)3B·(HF)n-catalyzed glycosylation of that donor, leading to efficient construction of the 1,2-trans-α-d-ido-heptopyransidic bonds through anchimeric assistance. In addition, 1,2-cis-α-d-galactopyranosidic linkages are highly stereoselectively achieved by employing the solvent effects of ether together with the capability of perchlorate counterions to favor α-stereoselectivity of glycosylation reaction. The accessibility of oligosaccharide 7 sets a solid foundation for glycoconjugate vaccine development. The established strategy for the construction of glycosidic bonds would facilitate synthesis of related glycans arising from C. jejuni capsular polysaccharides.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful for financial support from the Marine S & T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2022QNLM030003-2), the National Natural Science Foundation of China (Nos. 21977088 and 21672194), the National Natural Science Foundation of China-Shandong Joint Foundation (No. U1906213).
Supplementary material associated with this article can be found, in the online version, at doi:
G.V. Lopes, T. Ramires, N.R. Kleinubing, et al., Microb. Pathog. 161 (2021) 105265. doi: 10.1016/j.micpath.2021.105265
S. Kuwabara, Curr. Neurol. Neurosci. Rep. 7 (2007) 57–62. doi: 10.1007/s11910-007-0022-6
J.E. Pope, A. Krizova, A.X. Garg, Semin. Arthritis Rheum. 37 (2007) 48–55. doi: 10.1016/j.semarthrit.2006.12.006
K.O. Poropatich, C.L.F. Walker, R.E. Black, J. Health. Popul. Nutr. 28 (2010) 545–552.
Diarrhoeal Disease. World Health Organization, May 2, 2017.
K.U. Jansen, C. Knirsch, A.S. Anderson, Nat. Med. 24 (2018) 10–20. doi: 10.1038/nm.4465
J. Zhao, G. Hu, Y. Huang, Chin. Chem. Lett. 32 (2021) 1331–1340. doi: 10.1016/j.cclet.2020.10.013
P. Anderson, R.A. Insel, D.H. Smith, et al., J. Infect. Dis. 144 (1981) 530–538. doi: 10.1093/infdis/144.6.530
A.V. Karlyshev, D. Linton, N.A. Gregson, et al., Mol. Microbiol. 35 (2000) 529–541.
E.K. Jagusztyn-Krynicka, P. Łaniewski, A. Wyszyńska, Expert Rev. Vaccines 8 (2009) 625–645. doi: 10.1586/erv.09.21
F. Poly, A.J. Noll, M.S. Riddle, et al., Hum. Vaccines Immunother. 15 (2019) 1389–1400. doi: 10.1080/21645515.2018.1528410
M.A. Monteiro, S. Baqar, E.R. Hall, et al., Infect. Immun. 77 (2009) 1128–1136. doi: 10.1128/IAI.01056-08
Z. Pakulski, F. Poly, N. Dorabawila, et al., Curr. Org. Chem. 18 (2014) 1818–1845. doi: 10.2174/1385272819666140527231937
M. Cloutier, C. Gauthier, ACS Infect. Dis. 7 (2021) 969–986. doi: 10.1021/acsinfecdis.0c00332
T. Li, J. Wang, X. Zhu, et al., J. Am. Chem. Soc. 143 (2021) 11171–11179. doi: 10.1021/jacs.1c05048
W. Song, J. Cai, X. Zou, et al., Chin. Chem. Lett. 29 (2018) 27–34. doi: 10.1016/j.cclet.2017.09.061
M.A. Monteiro, A. Noll, R.M. Laird, et al., Campylobacter jejuni capsule polysaccharide conjugate vaccine, in: A.K. Prasad (Ed. ), Carbohydrate-Based Vaccines: From Concept to Clinic, American Chemical Society, 2018, pp. 249–271.
S. Zhang, P.H. Seeberger, Chem. Eur. J. 27 (2021) 17444–17451. doi: 10.1002/chem.202103234
P. Cmoch, A. Korda, L. Rárová, et al., Eur. J. Org. Chem. 19 (2014) 4089–4098.
X. Zhou, H. Ding, P. Chen, et al., Angew. Chem. Int. Ed. 59 (2020) 4138–4144. doi: 10.1002/anie.201914557
H. Ding, P. Wang, M. Li, Period. Ocean Univ. China 50 (2020) 95–99.
Q. Long, J. Gao, N. Yan, et al., Org. Chem. Front. 8 (2021) 3332–3341. doi: 10.1039/D1QO00211B
B. Schumann, H.S. Hahm, S.G. Parameswarappa, et al., Sci. Transl. Med. 9 (2017) eaaf5347. doi: 10.1126/scitranslmed.aaf5347
L. Cipolla, L. Lay, F. Nicotra, J. Org. Chem. 62 (1997) 6678–6681. doi: 10.1021/jo970127f
F. Nicotra, L. Panza, G. Russo, J. Org. Chem. 52 (1987) 5627–5630. doi: 10.1021/jo00234a024
E.K. Pathan, B. Ghosh, A.R. Podilapu, et al., J. Org. Chem. 86 (2021) 6090–6099. doi: 10.1021/acs.joc.0c02935
I. Tvaroŝka, T. Bleha, Adv. Carbohyd. Chem. Biochem. 47 (1989) 45–123.
P.I. Abronina, A.I. Zinin, N.N. Malysheva, et al., Synlett 28 (2017) 1608–1613. doi: 10.1055/s-0036-1589028
E. Eby, C. Schuerch, Carbohydr. Res. 34 (1974) 79–90. doi: 10.1016/S0008-6215(00)80372-9
H. Jona, H. Mandai, W. Chavasiri, et al., Bull. Chem. Soc. Jpn. 75 (2002) 291–309. doi: 10.1246/bcsj.75.291
R. Arihara, K. Kakita, K. Yamada, et al., J. Org. Chem. 80 (2015) 4278–4288. doi: 10.1021/acs.joc.5b00139
S. van der Vorm, T. Hansen, H.S. Overkleeft, et al., Chem. Sci. 8 (2017) 1867–187. doi: 10.1039/C6SC04638J

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



