
Scheme 1.
Schematic illustration of (A) design of MSCs-(MET+PTX) nanoplatform and (B) its antitumor mechanism.
Leveraging nano-engineered mesenchymal stem cells for intramedullary spinal cord tumor treatment
Lu Tang , Mengying Xie , Jing Li , Yijun Mei , Yuqi Cao , Qiaqia Xiao , Haijuan Dong , Yuhui Zhang , Wei Wang

Intramedullary spinal cord tumor (IMSCT) are relatively rare malignant tumors in the central nervous system and usually develop without obvious symptoms at the early stage [1]. When they can be medically detected, the tumors have already grown to a considerable size, which compresses the spinal cord in the marrow, leading to neurological dysfunction or mortality [2]. Currently, surgery is the preferred method to treat IMSCT in clinic, however, surgical operation is often very difficult due to the specific location, small size and invasiveness of IMSCT [3]. If the excision is too extensive, the dysfunction of spinal cord will lead to paralysis, incontinence and other serious consequences. If the resection range is small, the residual tumor cells infiltrating into the spinal parenchyma will cause the recurrence of IMSCT [4]. Although radiotherapy and chemotherapy play a vital role in antitumor treatment, radiation is likely to aggravate spinal cord injury, while the therapeutic efficacy of chemotherapy is often unsatisfactory because of the limited blood supply in the spinal canal [5, 6]. Due to the rareness of IMSCT compared to other tumor types, very little researches have been carried out to address this malignant tumor from the aspect of nanomedicine, which is yet very important for the management of IMSCT.
In light of this, our work firstly reported a targeted strategy by leveraging nano-engineered mesenchymal stem cells (MSCs) to carry therapeutic agents for IMSCT treatment. MSCs have been widely employed as cell-based vectors in antitumor therapy due to their low immunogenicity, tumor tropism and penetrability [7]. Compared to embryonic stem cells, MSCs are much less likely to differentiate to tumors after transplantation [8]. Moreover, MSCs have been clinically used to treat spinal cord injury, providing a basis for its application in IMSCT treatment [9]. In general, MSCs can deliver chemotherapeutic agents by carrying drug-loaded nanoparticles, which not only attenuates the direct cytotoxicity towards MSCs, but also combines the unique property of nanoparticles with the excellent tumor homing ability of MSCs [10]. Compared with internalized by MSCs, attachment of nanoparticles onto the surface of MSCs can further compromise the overall toxicity of nanomaterials to MSCs, preserve the integrity of nanoparticles and avoid the possibility that internalized nanoparticles cannot be expelled from the cells [11-13]. Studies have already demonstrated that nanoparticle-conjugated MSCs could still remain cell viability and tumor tropism, and there are many natural functional groups existing on the cell surface that can be covalently bonded with nanoparticles [14, 15].
In this work, maleimide-modified poly(lactic-co-glycolicacid) (PLGA-MAL) nanoparticles were synthesized to co-load therapeutic agent paclitaxel (PTX) and metformin (MET), which were then covalently attached onto MSCs surface via the thioether bond formed between the maleimide groups on PLGA-MAL and the thiol groups on cell membrane [16-19]. Although MET is a classic hypoglycemic drug, recent studies have demonstrated the antitumor efficacy of MET by activating adenosine monophosphate-activated protein kinase (AMPK) signaling pathway, which directly inhibits tumor cell growth [20, 21]. Furthermore, MET was reported to sensitize tumor cells towards chemotherapeutic agent such as PTX, doxorubicin and cisplatin [22]. Therefore, co-delivery of PTX and MET using PLGA-MAL nanoparticles will not only produce enhanced antitumor effectiveness than single drug treatment, but also cause less adverse effects compared to free drug administration. After intravenous injection, the prepared nanoparticle-conjugated MSCs could migrate towards IMSCT sites and penetrate into the deep tumor tissue due to the excellent homing ability and penetrability of MSCs. Afterwards, MET and PTX were continuously released around the tumor site because of the good biodegradability and sustained release behavior of PLGA, exerting long-term synergistic antitumor effect (Scheme 1).
Firstly, PLGA-MAL was synthesized according to the synthesis route shown in Fig. S1A (Supporting information), PLGA-OH was modified with maleimide through the condensation reaction between the hydroxyl group of PLGA-OH and the succinimide group of BMPS, which enabled its further attachment onto MSCs surface via the thioether bond formed between PLGA-MAL and MSCs. The successful synthesis of PLGA-MAL and its chemical structure was characterized by 1H NMR, FT-IR and UV spectra (Figs. S1B–D in Supporting information). As the 1H NMR of PLGA-MAL indicated, the characteristic proton peaks of PLGA-OH (δ 1.48–1.50 ppm and 4.10–5.35 ppm), and the proton peaks of maleimide group (δ 6.74 ppm), CH2N (δ 3.92 ppm) and CH2=O (δ 3.01 ppm) of BMPS all appeared [23, 24]. Moreover, the proton peaks of NHS group in BMPS disappeared in the 1H NMR spectrum of PLGA-MAL, implying the successful synthesis of PLGA-MAL. In FT-IR spectrum of PLGA-MAL, the peak of νC=O appeared at 1757 cm−1, which was similar to that in PLGA-OH because of the dominated long carbon chain of PLGA in PLGA-MAL. The peaks appeared at 2997–2882 cm−1 in PLGA-MAL belonged to the νC-H of CH3 and CH2 in PLGA-OH. Besides, the peaks at 3514 and 3652 cm−1 in PLGA-MAL belonged to the νC-H of CH2 from BMPS, which showed a chemical shift compared to the νC-H of CH2 in BMPS (3460 and 3505 cm−1), indicating the formation of ester bond in PLGA-MAL. Furthermore, as the UV spectra demonstrated, BMPS and PLGA-OH had absorbance peaks at 215 nm and 209 nm, respectively. PLGA-MAL also had an absorbance peak at 215 nm with the similar peak shape as BMPS, implying that BMPS was successfully conjugated to PLGA-OH.
PLGA-MAL/(MET+PTX) nanoparticles were fabricated by a double emulsion solvent evaporation method (Scheme S1 in Supporting information). The hydrodynamic diameter of these nanoparticles were 245.9 ± 3.3 nm with a narrow size distribution and their zeta potential was -38.41 ± 0.10 mV in average, which was consistent with the morphology observed by TEM (Fig. 1A, Figs. S2 and S3 in Supporting information). Meanwhile, SEM image demonstrated the uniform size of PLGA-MAL/(MET+PTX) nanoparticles with a smooth surface, which showed a milky white appearance in its suspension (Fig. 1B). Differential scanning calorimeter (DSC) technique was also performed to characterize the prepared nanoparticles (Fig. S4 in Supporting information). The encapsulation efficiency (EE) of PTX and MET were 84.40% ± 2.98% and 42.48% ± 0.06%, respectively, demonstrating their efficient loading in prepared nanocarrier (Figs. S5 and S6 in Supporting information). In vitro serum stability of PLGA-MAL/(MET+PTX) nanoparticles was also evaluated to investigate whether the fabricated nanoparticles could maintain stable in circulation. Although there was a fluctuation of the particle size during incubation with 10% FBS at 37 ℃ for 120 h, the variation of particle size was within 20% with narrow PDI, indicating the stability of nanoparticles during in vivo transportation (Fig. 1C). Additionally, in vitro drug release behavior of PTX and MET from prepared nanoparticles was determined (Fig. 1D). A burst release of both PTX and MET could be achieved in the first 24 h, while a more sustained release behavior was observed during the 14-day period for both drugs due to the biodegradability of PLGA [25]. Notably, the drug release rate of both MET and PTX was faster in medium at pH 6.8 than that at pH 7.4. The cumulative release of PTX measured by HPLC was approximately 80% on day 14 and nearly all the MET was released from the nanocarrier within 120 h at pH 6.8, implying that the designed nanoparticles were stable in physiological condition and could achieve effective drug release in the acid tumor microenvironment [26].
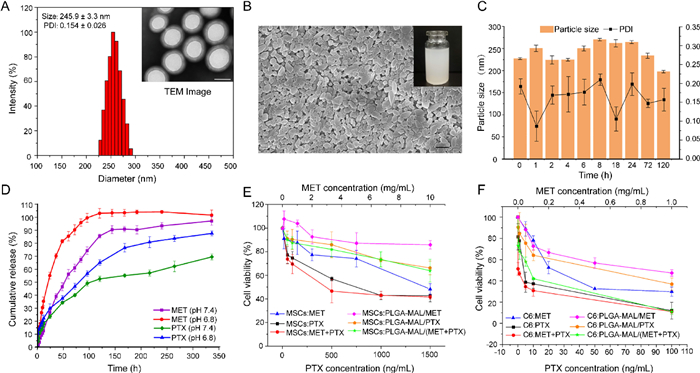
A drug nanocarrier that can effectively conjugate with MSCs without obvious cytotoxicity towards MSCs is important for MSCs-based cyto-pharmaceuticals. Therefore, MTT assay was conducted to determine the cytotoxicity of both free drugs and prepared formulations towards MSCs and C6 glioma cells (Figs. 1E and F). The cell viability of MSCs after treatment with high concentrations of free PTX (50–1500 ng/mL) and MET (0.1-10 mg/mL) still maintained a relatively high level, while just low concentrations of free PTX (0.1–100 ng/mL) and MET (0.05–1 mg/mL) could cause obvious cytotoxicity towards C6 cells, indicating that MSCs have a better tolerance of PTX and MET than C6 cells [27]. Moreover, the IC50 values of PTX and MTX towards MSCs and C6 cells were calculated, which showed that the IC50 of PTX and MET towards MSCs was 156 times and 12 times higher than C6 cells, respectively, demonstrating the stronger cytotoxicity of both therapeutic agents towards C6 cells compared with MSCs. Interestingly, the IC50 value in C6 cells after co-treatment with PTX and MET was significantly decreased by 10-fold compared to single PTX, while no similar phenomenon was observed in MSCs, implying that MET could increase the sensitivity of tumor cells towards PTX without affecting the viability of MSCs (Table S1 in Supporting information). Moreover, MSCs treated with wide-ranging doses of PLGA-MAL-based formulations maintained relatively high levels of viability, while an obvious dosage-dependent cytotoxicity was observed in C6 cells, further indicating the good tolerance of MSCs to drug-loaded nanoparticles. Thus, the above observations all proved that MSCs are ideal cell-based vectors and can tolerate the released free drugs at tumor sites.
To verify the formation of thioether bonds between MSCs and PLGA-MAL/(MET+PTX) nanoparticles, the availability of free thiol groups on MSCs surface was assessed by labeling them with F5M [28, 29]. Confocal images of F5M-labeled MSCs clearly demonstrated the presence of thiol groups on the surface of MSCs, which distributed uniformly over the entire cell surface (Fig. S7A in Supporting information). The conjugation of nanoparticles with MSCs was evaluated by co-incubating fluorescently tagged PLGA-MAL/FITC nanoparticles with MSCs, which was then analyzed quantitatively by flow cytometry and observed using inverted fluorescence microscope. In order to avoid the influence of cell adherence on the conjugation with nanoparticles, both adherent MSCs and suspended MSCs were used to co-incubate with prepared nano-formulations. As is shown in Figs. S7B and C (Supporting information), the surface attachment of nanoparticles showed a concentration-dependent manner and reached to the maximum when co-incubation with 500 µg/mL nanoparticles both in adherent and suspended cells. Moreover, the concentration-dependent fluorescence increment was also observed in nanoparticle-treated MSCs (Fig. S7D in Supporting information). It is worth noting that most of the membrane-anchored nanoparticles located on the main body of MSCs, but were rarely observed on their dendrite-like long protrusions that related to cell migration, which implied that the conjugation of nanoparticles on MSCs might not affect their migration ability [30, 31]. The quantitative analysis of drugs loaded on the MSCs was calculated and shown in Fig. S8 (Supporting information).
In order to verify whether the loaded nanoparticles would affect the tumor tropism of MSCs, a standard transwell migration assay was performed in this study [32]. In comparison to the control group without C6 cells, MSCs exhibited a relatively higher migration ability in other groups in the presence of C6 cells, indicating the tumor homing ability of nanoparticle-conjugated MSCs (Fig. 2A). Notably, as the quantitative analysis demonstrated, although the migration ability of nanoparticle-conjugated MSCs was slightly weaker than that of untreated MSCs, there was no significant statistical difference, verifying the good migration ability of MSCs towards tumor sites after conjugation with nanoparticles. 3D in vitro models of cell culture was exploited to mimic the tumor microenvironment in a closer way, which fills the gap between the traditional 2D cell condition and the in vivo environment [33]. In this study, a 3D model of C6 cells with round, dense, and well-organized structure was established to investigate the tumor penetration ability of nanoparticle-conjugated MSCs in vitro. Nanoparticle conjugated-MSCs (termed as MSCs-(MET+PTX)) labeled by FITC (green) could penetrate inside the tumor spheroid after 72 h incubation, which was nearly comparable to the penetrability of untreated MSCs labeled by DiI (red). In contrast, only very little amount of free nanoparticles could reach into the deep tumor spheroid (Fig. 2B). Therefore, all the results demonstrated that nanoparticle-conjugated MSCs could reserve the tumor penetration ability of untreated MSCs, which also exhibited superior penetrability over free nanoparticles.
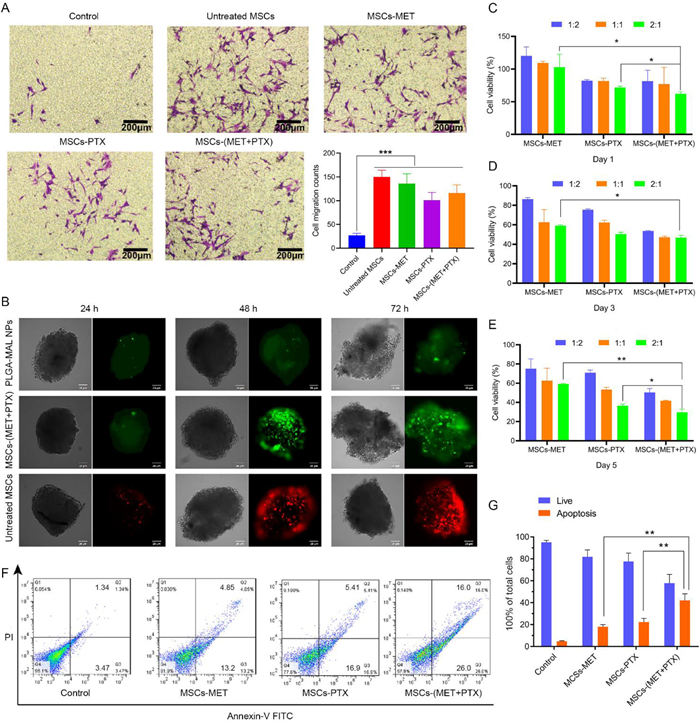
MTT assay was carried out to testify the antitumor potential of prepared MSCs-based nanoparticles in vitro. Transwell plates with the pore size of 0.4 µm were used to separate MSCs-based nanoparticles and C6 cells [34]. The cytotoxicity towards C6 cells after treatment with different MSCs-based nanoparticles exhibited a time- and dosage-dependent tendency (Figs. 2C–E). The most significant inhibitory effect on C6 cell growth with less than 30% cell viability could be observed after treatment with MSCs-(MET+PTX) for 5 days at the ratio of MSCs to C6 cells in 2:1, indicating the best antitumor efficacy due to the combination of PTX and MET. In addition, the apoptosis of C6 cells after 5-day treatment with MSCs-based nanoparticles was measured by Annexin V-FITC apoptosis assay and quantified by flow cytometry (Figs. 2F and G). In comparison to the control group, all the other groups treated with MSCs-based nanoparticles showed enhanced C6 cell apoptosis. Notably, MSCs-(MET+PTX) group caused the highest apoptosis ratio of around 42% towards C6 cells, further demonstrating the excellent therapeutic efficacy after co-treatment with MET and PTX.
All experiments involving animals were performed in strict accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and was approved by the Ethics Committee of China Pharmaceutical University (Ethics Code: 2021-12-002). The IMSCT model was established in rats to evaluate the antitumor efficacy in vivo, which was verified by H & E and immunohistochemistry staining (Figs. 3A–C and Fig. S9 in Supporting information). As the results of H & E staining indicated, there was an obvious tumor cell invasion in the spine of rat in surgery group compared to sham group, which was characterized by high cell density, diffuse cell growth with irregular morphology and clear lesion boundary, infiltrating and compressing the surrounding normal spinal cord tissue. Moreover, the biomarkers glial fibrillary acid protein (GFAP) and S100B that used as predictors for malignant glioma showed positive results (brown) in the immunostain of tumor-bearing spines [35]. Neuron-specific nuclear protein (NeuN) is often used as a neuronal biomarker in together with GFAP, which also exhibited positive effect with enlarged nucleus of tumor cells and obvious cytoplasm in deep staining [36]. Vimentin, a kind of major cytoskeletal proteins that related to cellular structure, is often used as a predictive biomarker for cancer. The immunostain of vimentin also appeared positive with yellowish-brown cytoplasmic staining [37, 38]. Altogether, the results above all demonstrated that C6 cells survived and developed into tumors in the spinal cord of rat.

The tumor targeting effect of MSCs-based nanoparticles was investigated in vivo using C6 tumor-bearing rats [39]. DiI was used as a fluorescent probe to track the in vivo distribution of MSCs-based nanoparticles. The biodistribution of DiI-labeled nanoparticles was detected by ex vivo imaging 5 days after injection. Compared with PLGA-MAL/DiI, DiI-labeled MSCs-(MET+PTX) could effectively accumulate in the tumor site like the untreated MSCs, both exhibiting strong fluorescence intensity in tumor tissue without significant difference. Moreover, the quantified analysis of the fluorescent signal in main organs and tumor tissue also verified the excellent tumor targetability of MSCs-(MET+PTX) compared to PLGA-MAL/DiI (Figs. 3D and E). As the fluorescent images of spinal cord tissue slides displayed in Fig. 3F, untreated MSCs and MSCs-(MET+PTX) group showed strong fluorescent signals of DiI, while negligible DiI signal could be observed in control group, further demonstrating the outstanding homing capability of MSCs-based nanoparticles towards spinal cord tissue and the surface attachment of nanoparticles on MSCs would not affect their migration ability.
IMSCT model was established to evaluate the in vivo antitumor efficacy of nanoparticle-conjugated MSCs. BBB locomotor rating scale that reflects the degree of paralysis by scoring the hindlimb movement of rats was used to assess the spinal cord injury due to glioma [40, 41]. In brief, C6 tumor-bearing rats were administrated with various MSCs-based nanoparticles on day 10 after the IMSCT model was set up according to the regimen shown in Fig. 4A. During 3-week treatment period, the body weight and hindlimb function of rat were recorded according to BBB score. As is shown in Fig. 4B and Fig. S10 (Supporting information), in comparison to the significant loss of body weight in control group without treatment, the body weight of rats administrated with various MSCs-based formulations increased during the treatment period, which was nearly closed to that of sham group, indicating the significant inhibitory effect on tumor growth after treatment. Furthermore, according to the BBB evaluation in Fig. 4C, treatment with MSCs-based formulations could remarkably improve the hindlimb movement of rats compared to the control group. Notably, the co-loading of MET and PTX on MSCs showed a significantly enhanced therapeutic efficacy than single drug. Meanwhile, as the Kaplan-Meier graph displayed in Figs. 4D and E, treatment with MSCs-(MET+PTX) could largely delayed the hindlimb paralysis of rats and improved their survival rates in comparison to the control group, and there was no rat with BBB score less than 5 after administration with MSCs-(MET+PTX).
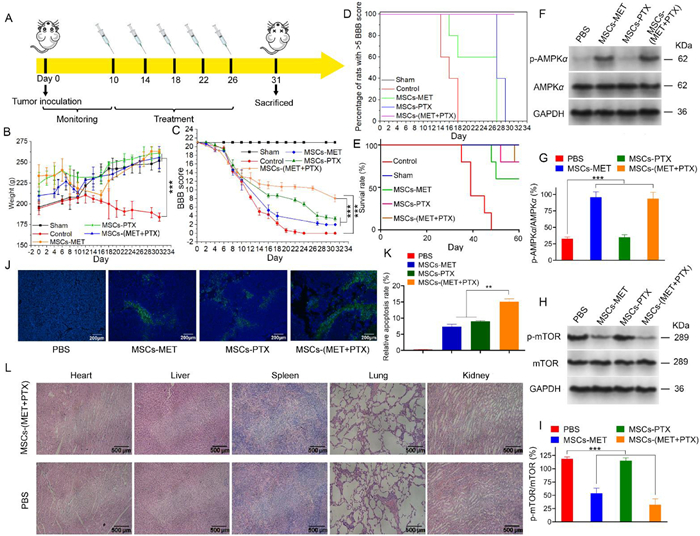
In addition, metformin was reported to inhibit tumor cells by activating the AMPK and inhibiting the mTOR pathways [42]. AMPK serves as a metabolic tumor suppressor that regulates glucose and lipid metabolism. A low phosphorylation level of AMPK is often associated with poor prognosis after cancer treatment [43]. Therefore, the expression levels of AMPKα, phosphorylated AMPKα, mTOR and phosphorylated mTOR in tumors after treatment with various MSCs-based formulations were checked through Western blotting assay. As shown in Figs. 4F–I, the phosphorylation levels of AMPKα in tumor sites after treatment with MSCs-MET and MSCs-(MET+PTX) groups were significantly increased compared to the PBS control and MSCs-PTX. Phosphorylation of mTOR plays an important role in tumor cell proliferation and survival [44]. Thus, the effects on the activity of mTOR after various treatment groups were also assessed. Treatment with MSCs-MET and MSCs-(MET+PTX) resulted in a significant inhibitory effect on mTOR activity evidenced by stronger reduction in p-mTOR/mTOR levels compared with the PBS group and MSCs-PTX.
TUNEL staining of the spinal cord sections of C6 tumor-bearing rats showed that there was obvious tumor cell apoptosis in rats treated with MSCs-based formulations compared to the PBS control, indicating the great therapeutic efficacy of prepared formulations (Figs. 4J and K). Meanwhile, a significantly enhanced apoptosis rate was identified in MSCs-(MET+PTX) group compared to MSCs-MET and MSCs-PTX groups, which verified the best therapeutic performance of the combination application of MET and PTX. In vivo safety of MSCs-(MET+PTX) was also assessed after 3-week treatment. As the H & E staining presented in Fig. 4L, there was no obvious abnormality in the morphology of major organs after treatment with MSCs-(MET+PTX) compared to the PBS control, indicating that the prepared formulations would not cause apparent damage to the body and have good biosafety. Moreover, the serum level of ALT, AST and BUN was determined in normal rats treated with MSCs-(MET+PTX) after 3 weeks to evaluate the liver and renal function, which also showed no obvious change compared to the PBS control (Fig. S11 in Supporting information) [45, 46]. Based on these results, it can be concluded that the fabricated MSCs-(MET+PTX) platform possessed excellent therapeutic efficacy against spinal glioma with good targetability and satisfactory in vivo safety, exhibiting valuable research potential for IMSCT treatment.
In summary, this work presented a potential strategy based on co-delivery of MET and PTX by nano-engineered MSCs for IMSCT treatment. Due to the fact that there are very limited reported researches for targeted IMSCT therapy using nanomedicine, our work originally leveraged MSCs to deliver therapeutic agents towards difficult accessible IMSCT sites owing to the great tumor tropism of MSCs, providing more opportunities for precise IMSCT management beyond surgery. The designed nano-engineered MSCs showed potent therapeutic efficacy against IMSCT with negligible toxicity in vivo by synergizing the antitumor effect of MET and PTX, meanwhile achieving sustained drug release due to the biodegradability of PLGA to enable long-term antitumor efficacy. With the expectation to exploit more alternatives for treating IMSCT, this work might offer a reasonable strategy for targeting IMSCT based on nanoparticle-conjugated MSCs.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Nature Science Foundation of China (Nos. 31872756 and 32071387), and Six Talent Peaks Project in Jiangsu Province (No. JY-079), 333 High-level Talent Development Project in Jiangsu Province.
Supplementary material associated with this article can be found, in the online version, at doi:
M. Bydon, A. Goyal, P. Kerezoudis, World Neurosurg. 157 (2022) 123–124. doi: 10.1016/j.wneu.2021.10.133
I. Yuce, O. Kahyaoglu, H.A. Cavusoglu, et al., J. Clin. Neurosci. 86 (2021) 26–31. doi: 10.1016/j.jocn.2021.01.001
I. Hussain, W.E. Parker, O. Barzilai, M.H. Bilsky, Neurosurg. Clin. N. Am. 31 (2020) 237–249. doi: 10.1016/j.nec.2019.12.004
W. Hsu, C. Bettegowda, G.I. Jallo, Childs Nerv. Syst. 26 (2010) 241–245. doi: 10.1007/s00381-009-1022-4
F. Siddiqui, B. Movsas, Semin. Radiat. Oncol. 27 (2017) 340-349. doi: 10.1016/j.semradonc.2017.04.008
T. Kutluk, A. Varan, C. Kafali, et al., Eur. J. Paediatr. Neurol. 19 (2015) 41–47. doi: 10.1016/j.ejpn.2014.09.007
Y. Takayama, K. Kusamori, M. Nishikawa, Expert Opin. Drug Deliv. 18 (2021) 1627–1642. doi: 10.1080/17425247.2021.1960309
S.S. Hakki, G. Turac, S.B. Bozkurt, et al., Cells Tissues Organs 204 (2017) 228–240. doi: 10.1159/000478998
S.L. Lindsay, G.A. McCanney, A.G. Willison, S.C. Barnett, Nat. Rev. Neurol. 16 (2020) 229–240. doi: 10.1038/s41582-020-0311-0
D. Dapkute, M. Pleckaitis, D. Bulotiene, et al., ACS Appl. Mater. Interfaces 13 (2021) 43937–43951. doi: 10.1021/acsami.1c10445
Q. Wang, H. Cheng, H. Peng, et al., Adv. Drug Deliv. Rev. 91 (2015) 125–140. doi: 10.1016/j.addr.2014.12.003
A.C. Anselmo, J.B. Gilbert, S. Kumar, et al., J. Control. Release 199 (2015) 29-36. doi: 10.1016/j.jconrel.2014.11.027
L. Tang, S. He, Y. Yin, et al., Pharmaceutics 13 (2021) 1888. doi: 10.3390/pharmaceutics13111888
E.C. Wayne, S. Chandrasekaran, M.J. Mitchell, et al., J. Control. Release 223 (2016) 215–223. doi: 10.1016/j.jconrel.2015.12.048
B. Huang, W.D. Abraham, Y. Zheng, et al., Sci. Transl. Med. 7 (2015) 291ra294.
M.T. Stephan, S.B. Stephan, P. Bak, J. Chen, D.J. Irvine, Biomaterials 33 (2012) 5776–5787. doi: 10.1016/j.biomaterials.2012.04.029
X. Wang, H. Chen, X. Zeng, et al., Acta Pharm. Sin. B 9 (2019) 167–176. doi: 10.1016/j.apsb.2018.08.006
P. Huang, D. Lian, H. Ma, et al., Chin. Chem. Lett. 32 (2021) 3696–3704. doi: 10.1016/j.cclet.2021.06.034
H. Wang, L. Wang, Y. Gao, Y. Ding, Chin. Chem. Lett. 32 (2021) 1041–1045. doi: 10.1016/j.cclet.2020.08.044
R. Ma, B. Yi, A.I. Riker, Y. Xi, Acta Pharmacol. Sin. 41 (2020) 1403–1409. doi: 10.1038/s41401-020-00508-0
J.H. Cha, W.H. Yang, W. Xia, et al., Mol. Cell 71 (2018) 606–620e7. doi: 10.1016/j.molcel.2018.07.030
Y. Xiao, S. Wang, Q. Zong, Z. Yin, AAPS PharmSciTech 19 (2018) 2395–2406. doi: 10.1208/s12249-018-1070-8
J. Emami, P. Maghzi, F. Hasanzadeh, et al., Pharm. Dev. Technol. 23 (2018) 41–54. doi: 10.1080/10837450.2017.1340950
T. Zou, F. Dembele, A. Beugnet, et al., J. Biotechnol. 214 (2015) 147–155. doi: 10.1016/j.jbiotec.2015.09.034
P.S. Wei, Y.J. Chen, S.Y. Lin, et al., Biomaterials 278 (2021) 121166. doi: 10.1016/j.biomaterials.2021.121166
L. Tang, Y. Mei, Y. Shen, et al., Int. J. Nanomed. 16 (2021) 5811–5829. doi: 10.2147/ijn.s321416
A. Bonomi, A. Silini, E. Vertua, et al., Stem Cell Res. Ther. 6 (2015) 155. doi: 10.1186/s13287-015-0140-z
A. Catala, M. Dzieciatkowska, G. Wang, et al., ACS Nano 15 (2021) 11789–11805. doi: 10.1021/acsnano.1c02864
O. Akinloye, R. Krishnamurthy, D. Wishart, G.G. Goss, Anal. Bioanal. Chem. 409 (2017) 903–915. doi: 10.1007/s00216-016-0042-7
S. Seetharaman, S. Etienne-Manneville, Trends Cell Biol. 30 (2020) 720–735. doi: 10.1016/j.tcb.2020.06.004
A. Bisaria, A. Hayer, D. Garbett, D. Cohen, T. Meyer, Science 368 (2020) 1205–1210. doi: 10.1126/science.aay7794
K. Chen, X. Cao, M. Li, et al., Theranostics 9 (2019) 2984–2998. doi: 10.7150/thno.31157
M.H. Macedo, A. Baiao, S. Pinto, et al., Adv. Drug Deliv. Rev. 178 (2021) 113993. doi: 10.1016/j.addr.2021.113993
J. Lin, S. Cao, Y. Wang, et al., J. Exp. Clin. Cancer Res. 37 (2018) 113. doi: 10.1186/s13046-018-0727-1
F. Galland, M. Seady, J. Taday, et al., Neurochem. Int. 131 (2019) 104538. doi: 10.1016/j.neuint.2019.104538
L. Zhang, Y. Guo, H. Hu, et al., Int. J. Med. Sci. 12 (2015) 288–294. doi: 10.7150/ijms.10527
J. Zhao, L. Zhang, X. Dong, et al., Appl. Immunohistochem. Mol. Morphol. 26 (2018) 337–344. doi: 10.1097/pai.0000000000000420
W. Lu, J.Y. Luo, M.H. Wu, et al., Pathol. Res. Pract. 215 (2019) 1020–1032. doi: 10.1016/j.prp.2019.02.010
T. Lan, M. Luo, X. Wei, J. Hematol. Oncol. 14 (2021) 195. doi: 10.1186/s13045-021-01208-w
D.M. Basso, M.S. Beattie, J.C. Bresnahan, J. Neurotrauma 12 (1995) 1–21. doi: 10.1089/neu.1995.12.1
R. Jian, Y. Yixu, L. Sheyu, et al., J. Biomed. Mater. Res. A 103 (2015) 3259–3272. doi: 10.1002/jbm.a.35466
R.J. Dowling, M. Zakikhani, I.G. Fantus, M. Pollak, N. Sonenberg, Cancer Res. 67 (2007) 10804–10812. doi: 10.1158/0008-5472.CAN-07-2310
E. Zulato, F. Bergamo, A. De Paoli, et al., Br. J. Cancer 111 (2014) 25–32. doi: 10.1038/bjc.2014.274
S. Matsubara, Q. Ding, Y. Miyazaki, et al., Sci. Rep. 3 (2013) 3230. doi: 10.1038/srep03230
L. Xu, Y. Yu, R. Sang, et al., Oxidative Med. Cell Longev. 2018 (2018) 8284107.
R.S. Loomba, E.G. Villarreal, J. Dhargalkar, et al., J. Clin. Pharm. Ther. 47 (2021) 287–297.

Scheme 1 Schematic illustration of (A) design of MSCs-(MET+PTX) nanoplatform and (B) its antitumor mechanism.

Figure 1 Characterization of the prepared nanoparticles. (A) Size distribution histogram, PDI and TEM image (insert) of PLGA-MAL/(MET+PTX) nanoparticles. Scale bar: 200 nm. (B) SEM image of PLGA-MAL/(MET+PTX) nanoparticles. Scale bar: 2 µm. Inset: PLGA-MAL/(MET+PTX) suspension. (C) Particle size and PDI of PLGA-MAL/(MET+PTX) nanoparticles after incubation with 10% FBS at different time points at 37 ℃. (D) In vitro release profiles of MET and PTX from PLGA-MAL/(MET+PTX) nanoparticles in PBS (pH 7.4 and 6.8) containing 0.5% Tween-80. (E) Cell viability of MSCs and (F) C6 cells treated with different formulations for 72 h. Data were presented as mean ± S.D. (n = 5).

Figure 2 (A) Representative images and quantification of migrated MSCs after exposure to different formulations in response to C6 cells. Scale bar: 200 µm. (B) 3D laser scanning confocal microscope images of C6 glioma spheroids. C6 glioma spheroids were incubated with PLGA-MAL/(MET+PTX) nanoparticles (FITC, green), MSCs-(MET+PTX) (FITC, green) and untreated MSCs (DiI, red) for 24, 48 and 72 h. Scale bar: 25 µm. (C–E) Cell viability of C6 cells exposed to MSCs-MET, MSCs-PTX and MSCs-(MET+PTX) with different cell density and incubation time. Flow cytometry analysis (F) and (G) statistical analysis of apoptotic C6 cells induced by different MSCs-based formulations on day 5. Data were shown as mean ± S.D. (n = 5, P < 0.05, **P < 0.01, ***P < 0.001).

Figure 3 (A) Schematic illustration of the surgical operation to establish IMSCT. (B) H & E-stained sections from the spines of sham-operated rats and surgery rats. Scale bar: 100 µm. (C) Immunohistochemical staining of the C6 tumor-bearing spines. Scale bar: 100 µm. Accumulation of DiI in tumor sites and major organs (D) detected by ex vivo imaging and (E) quantitative analysis on day 5 post-injection of different formulations. (F) In vivo migration of MSCs in IMSCT slides observed under invert fluorescence microscope. Data were shown as mean ± S.D., n = 5, ***P < 0.001.

Figure 4 (A) Treatment regimen. (B) Body weight analysis and (C) BBB scores of sham-operated and C6 tumor-bearing rats after treatment with PBS and different MSCs-based formulations. (D) Kaplan-Meier graph of the onset of hindlimb paralysis (BBB functional score < 5). (E) Survival curves of sham-operated rates and C6-tumor bearing rats treated with PBS and different MSCs-based formulations. (F, H) Representative western blot analysis of protein levels in C6 tumor sites after treatment with PBS and different MSCs-based formulations. GAPDH was used as loading control. (G, I) Quantitative analysis of the protein levels of phosphorylated AMPKα, AMPKα, phosphorylated mTOR, mTOR that are associated with the activation of AMPK pathway. (J) TUNEL staining and its (K) quantification of IMSCT sections in rats treated with PBS and different MSCs-based formulations. Scale bar: 200 µm. (L) H & E stained sections of major organs in rats treated with PBS and MSCs-(MET+PTX). Scale bar: 500 µm. Data were shown as mean ± S.D., n = 5, **P < 0.01, ***P < 0.001.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: