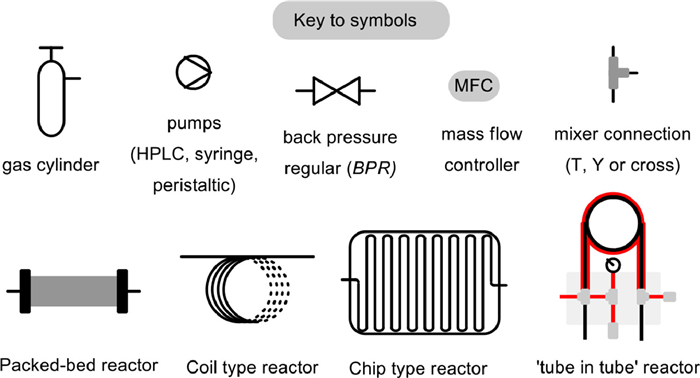
Figure 1.
Schematic diagram of reaction device. Reproduced with permission [42]. Copyright 2019, Wiley-VCH.
Recent advances in chemical fixation of CO2 based on flow chemistry
Hui Luo , Jing Ren , Ying Sun , Yunlin Liu , Feng Zhou , Guoyue Shi , Jian Zhou

Since pre-industrial times, the concentration of CO2 in atmosphere has gradually increased due to the combustion of fossil fuels to meet the world's energy demand. This increase is generally considered to be the main reason for the rise in atmospheric temperature and probably abnormal changes in global climate [1,2]. During the past decades, great efforts have been made to reduce the net amount of anthropogenic CO2 released in the atmosphere [3,4]. Among which, the development of chemical processes using CO2 as a promising alternative carbon-based C1 feedstocks of fossil fuel resource for the production of industrially attractive chemicals has received considerable attention [5-12]. Although the chemical fixation of CO2 cannot solve the global warming problem alone, it could provide access to materials of commercial interest from such an abundant, nontoxic, renewable, and low-cost carbon resource, thus offers interesting opportunities for the chemical industry, organic synthesis and so on [13,14]. However, as the end product of all carbon-based combustion process and possessing the highest oxidation state of carbon, CO2 is thermodynamically stable and kinetically inert [15,16]. To power the conversion of CO2 to value-added chemicals, harsh reaction conditions of high temperature and high pressure are often required, which induces the indirect production of additional CO2 thus might offset the beneficial effects of CO2 transformation to chemicals [17-19]. In this context, the development of novel system to enable the reaction under mild conditions is highly desirable.
Recent advances in flow chemistry provide effective means for the chemical transformation of CO2 and its incorporation into valuable organic compounds, and it has attracted much attention from both industry and academia due to their several well-defined advantages [20-25]. At first, the high surface area to volume ratio of flow reactor not only permits sufficient mixing of different phases, such as the gas CO2 with liquid reaction system or immobilized catalyst, but also enables precise temperature control. Such high mass and heat transfer rate could increase the reaction efficiency significantly, and also improve the process control effectively [26-28]. Secondary, the excellent thermal management and the low material hold-up in flow reactor could make its implementation much safer in practice, thus enabling transformations that are difficult, or simply not feasible to perform in batch, and also facilitating the rapid reaction optimization and streamline multistep processes [29-31]. Thirdly, the numbering-up methodology for its production throughput increase enables small flow reactors to produce comparably large quantities of product, thus largely reduces the time from lab research to industrial application [32].
Despite these advantages, the application of flow chemistry on CO2 transformation remains a challenging task. Due to the inherent low reactivity of CO2, many batch reactions often quite long reaction times, but it is usually not amenable to flow conditions as the resulting low flow rate might lead to inefficient mixing. Moreover, depending on the operating conditions and reactor geometry, different flow patterns such as slug flow, annular flow, bubble flow and churn flow might be formed, which influence the experimental results directly. In this context, the operative parameters of a continuous process will most probably differ substantially from a batch process, and need to be accurately adjusted [26]. On the other hand, the possible formation of carboxylic salt precipitation might lead to irreversible blocking of the flow reactor, thus represents another obstacle in the implementation of batch reactions into continuous flow. Furthermore, other problems such as the catalyst recycling, product separation as well as the flow rate synergy, solvent and/or waste compatibility for multistep reactions should also be considered for CO2 transformation under flow conditions.
Along with the rapid development of transformations employing CO2 as a C1 synthon in batch conditions [33-41], reactions with CO2 in continuous flow systems has attracted more and more attentions. Moreover, with the generation of new concepts, more efficiency catalysts, and novel equipments, innovative approaches have been reported for solving the above-mentioned challenges. Despite ongoing progress, the only one comprehensive review in this important and active research field was given by Jamison and co-workers in the beginning of 2019 [42]. Since then, many new methods and techniques that fully utilized the advantages of continuous flow platforms for the chemical fixation of CO2 have been realized. Multistep continuous synthesis of complex drugs using CO2 as C1 synthon have also emerged. In view of the rapid development and the urgent need for continuous transformation of CO2, herein we wish to present an update of the recent advances in this direction. We will divide the literature reports by the reaction types: the CO2 participated cycloaddition and carboxylation reactions. In each part, we will focus on the development of new catalysts and equipments in flow chemistry. While the possible mechanism, the synthetic utilization and the remaining challenges of current transformations will also be discussed. We hope this concise review can give inspiration to related researchers that how competitive are these flow chemistry-based approaches for the construction of valuable chemicals using CO2 as C1 synthon. Notably, flow chemistry should be seen as a powerful complementary tool to assist modern synthesis programs, rather than a direct competitor to batch chemistry. The schematic representations of some common equipment that may be implemented in a flow platform are illustrated in Fig. 1.
The cycloaddition of CO2 with epoxides to cyclic carbonates represents one of the most important reactions utilizing CO2 as a chemical feedstock [43,44]. As an important class of compounds featuring unique physical and chemical properties, cyclic carbonates have been frequently associated with numerous applications involving as environmentally friendly nonprotic solvents, electrolytes in batteries, raw materials for plastics, precursors for pharmaceutical intermediates and so on [45]. Although this reaction has been developed about sixty years ago [46], it is kinetically highly unfavorable due to the inherent high barriers, and usually requires relatively high temperature and pressure [47]. Therefore, the development of new catalytic systems as well as the refinement of flow reactors to improve the reaction efficiency and production capacity are highly desirable.
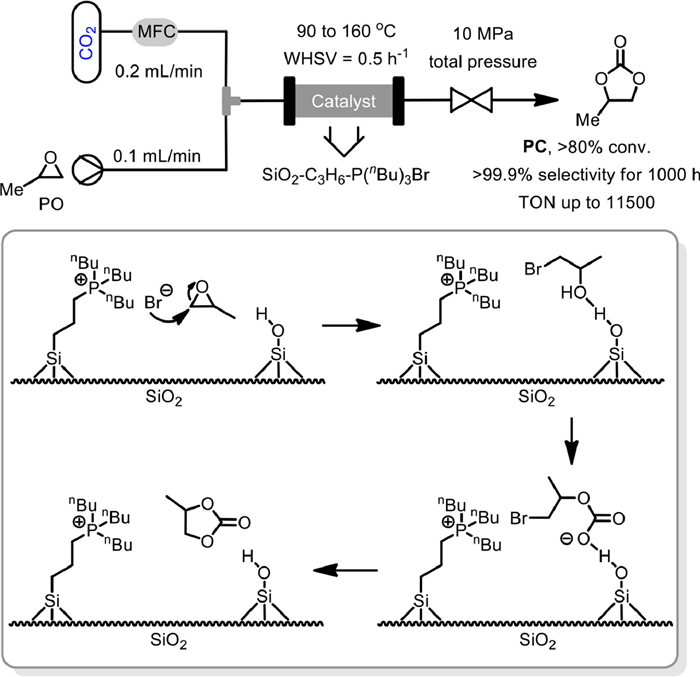
As early as 2006, Sakakura and co-workers reported the first cycloaddition of CO2 with propylene oxide (PO) to propylene carbonate (PC) in packed-bed reactor, by using SiO2 immobilized C3H6-P(n-Bu)3Br as a synergistic organic–inorganic hybrid catalyst (Scheme 1) [48]. Notably, the acidic inorganic silica support could not only facilitate catalyst separation, but also promote the catalytic activity of the basic phosphonium salt part. They found that the pseudo-first order rate constant of SiO2-C3H6-P(n-Bu)3I normalized to a phosphorus atom was about 300 times larger than that of P(n-Bu)4I. It was proposed that the acidic surface silanol groups could interact and activate propylene oxide, thus playing a synergistic role with halogen anion to promote the cycloaddition. Finally, with 10 g SiO2-C3H6-P(n-Bu)3Br (8.5 mmol phosphorus) as catalyst, the fixed-bed flow reactor experiment was performed under 10 MPa with a propylene oxide flow of 0.1 mL/min (weight hourly space velocity, WHSV = 0.5 h−1) and CO2 flow of 0.2 mL/min by gradually raising the temperature from 90 ℃ to 160 ℃. Finally, > 80% conversion of PO and > 99.9% selectivity could be achieved for more than 1000 h, with TON up to 11,500. This seminal research clearly demonstrated the powerfulness of packed-bed reactor and supported catalysts in CO2 cycloaddition to propylene oxide. From then on, a series of new supported catalysts were developed and successfully applied to the cycloaddition of CO2 with epoxides based on packed-bed reactors.
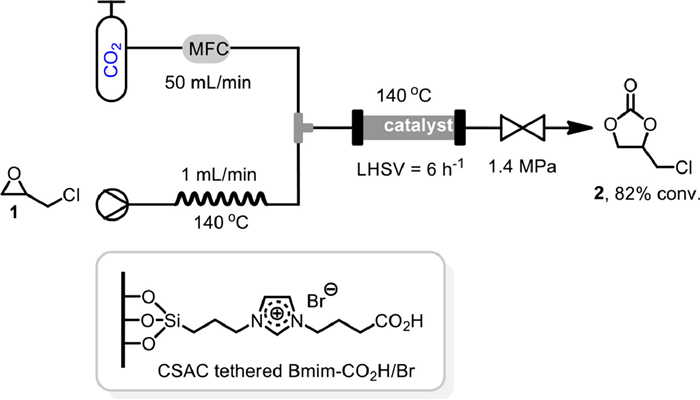
In 2015, Xiong et al. reported the cycloaddition of CO2 to epichlorohydrin 1 in a packed bed reactor under solvent free conditions, using coconut shell activated carbon (CSAC) tethered ionic liquids as catalyst (Scheme 2) [49]. Under 140 ℃ and 1.4 MPa, with the flow rates of epichlorohydrin 1 at 1 mL/min and CO2 at 50 mL/min, the CSAC tethered Bmim-COOH/Br could catalyze the continuous reaction to give a stabilized 82% conversion after 50 h, with liquid hourly space velocity (LHSV) of 6 h–1. It was proposed that the carboxyl group of the catalyst might form hydrogen bond with oxygen in epichlorohydrin 1 resulting in the polarization of C–O bond, which facilitated the following nucleophilic ring opening of Br− and CO2 insertion.
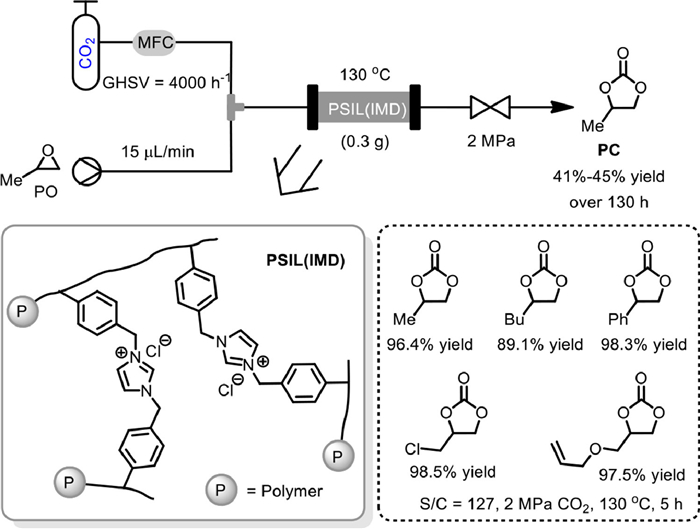
In 2017, Ding and co-workers synthesized a novel imidazolium-based polymer supported ionic liquids PSIL(IMD), via the solvothermal copolymerization of vinyl functionalized imidazolium chloride slat, and applied it to the flow conversion of CO2 to PC in fixed-bed reactor (Scheme 3) [50]. Under the catalysis of 0.3 g of PSIL(IMD), the PO was continuously fed (15 µL/min) without any solvents under 2 MPa of CO2, with gas hourly space velocity (GHSV) of 4000 h−1. During the study period of 130 h, PC could be obtained with a yield ranging between 41% and 45%, demonstrating the catalytic stability of PSIL(IMD). The performance of PSIL(IMD) under batch conditions was also studied, and various cyclic carbonates could be obtained in high yields with the S/C (substrate/catalyst) ratio of 127 under 2 MPa of CO2 at 130 ℃ for 5 h. These results further illustrated the high catalytic activity of the PSIL(IMD) catalyst.
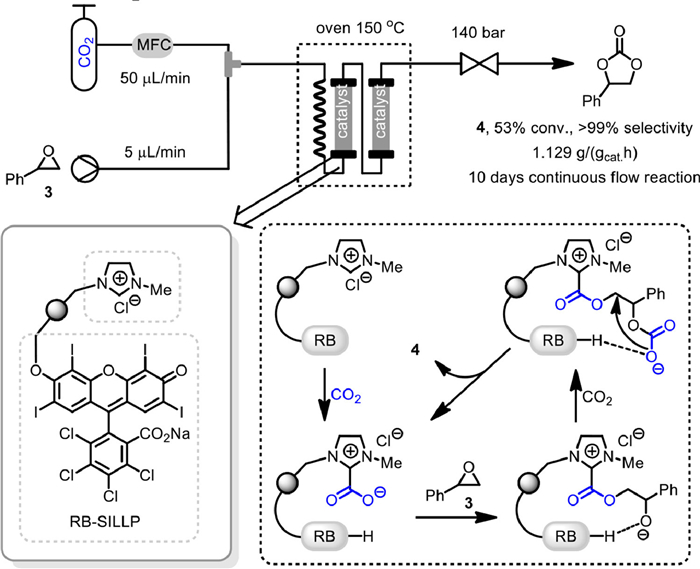
In 2021, García-Verdugo and Luis developed a novel type of multifunctional polymers based on ionic liquid and Rose Bengal (RB) fragments for the efficiently cycloaddition of CO2 and epoxide [51]. By adjusting the nature of the supported ionic liquid-like phases (SILLPs), the catalytic activity of RB fragments could be fine-tuned, thus proving high activity and stability during the continuous flow reaction conditions. As shown in Scheme 4, a preheater was employed to heating the mixture of CO2 (50 µL/min) and styrene oxide 3 (5 µL/min) before contacting the supported catalyst RB-SILLP. By fixing the reaction temperature at 150 ℃, and the total pressure of 140 bar, cyclic carbonate 4 can be obtained in 53% conversion and > 99% selectivity for 10 days, with a productivity of 1.129 gcyclocarbonate/(gcatalysth), among the higher productivities reported so far under continuous flow conditions. Moreover, no appreciable leaching of RB or decay on the catalytic activity was observed during the continuous operation. Based on systematic experiment studies it was proposed that the residual water and RB fragments could benefit the formation of NHC fragment from imidazolium unit. Then the reaction of NHC with CO2 gave zwitterionic NHC–CO2, which was considered to be the real catalytic species to promoted the following ring opening of epoxide and CO2 insertion steps.
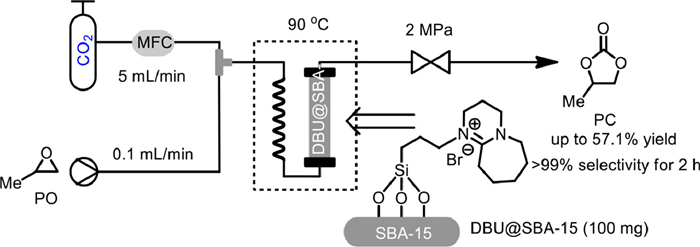
In 2021, a green and high-efficiency DBU-based ionic liquid catalyst immobilized on SBA-15, DBU@SBA-15, was reported by Yin and co-workers and applied to the cycloaddition of PO and CO2 to PC using packed bed reactors (Scheme 5) [52]. The cation [DBU]+ and anion [Br]− grafted with SBA-15 could catalyzed the cycloaddition in a synergistic manner. By performing the reaction under 2 MPa of CO2 at 90 ℃, with the PO flow rate of 0.1 mL/min, CO2 flow rate of 5 mL/min, 57.1% yield of PC could be achieved after continuous reaction for 2 hours. After 24 h of continuous use, 16.86% yield could also be maintained. But after 26 h, the catalyst activity was lost with only 4% yield obtained. However, the quality of the recovered catalyst was not leached out after continuous use for 30 h, demonstrating the stability of the catalyst. It was proposed that the deterioration of the catalytic activity might be caused by the occupation of active sites on the active center for a long time by CO2, and adding co-gas or desorption of CO2 after a certain time were suggested to circumvent the deactivation.
Apart from supported ionic liquids, other novel heterogeneous catalysts are also developed and applied to the cyclic carbonate synthesis from CO2 and epoxides. In 2020, Bui, Konwar and Mikkola employed inexpensive mesoporous melamine-formaldehyde resins MMFR as efficient heterogeneous catalyst for direct synthesis of cyclic carbonates from CO2 and epoxides [53]. As shown in Scheme 6, the continuous-flow reaction was performed in a down flow vertical fixed-bed reactor packed with the polymeric MMFR (1.95 g, ≤100 µm). Epoxides 5 was pumped into the reactor at a flow rate of 0.01-0.04 mL/min, with weight hourly space velocity (WHSV) of 0.26-1.9 h–1, while CO2 was simultaneously supplied with 15 mL/min. By fixing the reaction temperature at 120 ℃, and the total pressure of 13 bar, the cyclic carbonates 6 could be obtained continuously with 76%–100% conversion and > 99% selectivity during 7–13 days of operation. Notably, although the catalytic activities decreased in batch mode, due to the potential N-active sites poisoning, they showed exceptional stability in continuous flow conditions. It was believed that the abundant Lewis basic N-sites of the melamine resins was critical for the high catalytic activities, as it could activate CO2 molecules effectively and then facilitated the ring opening of the epoxides. Based on CO2-temperature programmed desorption, the concentration of surface basic sites for MMFR was estimated to be 172 µmol/g, while the activation energies of CO2 desorption (strength of basic sites) were calculated to be 92.1 kJ/mol. The highly catalytic activity and stability of MMFR demonstrated their potential for industrial-scale applications.

Along with the exploitation of new catalysts, a lot of focus have also been given to the development of new types of flow reactors aiming to further improve the production efficiency [54,55]. In this regard, the development and application of microreactors for the chemical fixation of CO2 have received considerable attention. In general, microreactors possess controllable and very high specific surface area, thus especially suitable for the gas–liquid two-phase processes. Moreover, the utilization of microreactors provides opportunity for the application of homogeneous catalysis for the CO2 based chemical processes.
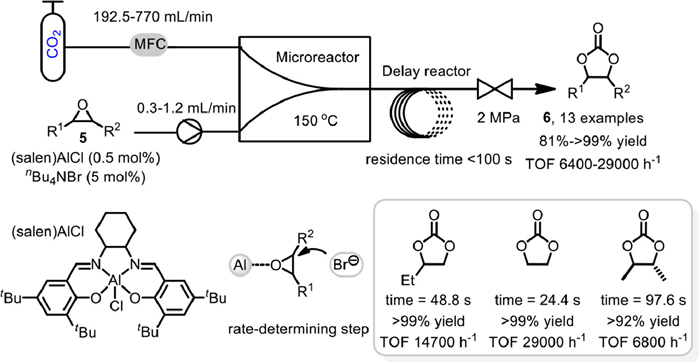
In 2018, Ren and co-workers reported the efficient coupling of CO2 with epoxides 5 to various cyclic carbonates 6 in microreactor under the catalysis of binary (salen)AlCl/nBu4NBr (TBAB) system (Scheme 7) [56]. The microreactor bearing a complex sloped structure, with geometric size of 300 µm × 300 µm × 1 cm (length) was employed as mixing channel, enabling the multiple separation and recombination of liquid and gas phases to complete the ample mixing process. After which, a delay reactor (1/8 inch 316L stainless steel tube, wall thickness 0.5 mm, 22.83 mL) was used for the reaction. By adjusting the flow rate of epoxides 5 and CO2 to change the retention time, a variety cyclic carbonates 6 could be obtained with high yields (> 90%) and excellent selectivity (99%) under 150 ℃ and 2.0 MPa CO2 pressure within the residence time of less than 100 s. Notably, compared to the reaction in a conventional stirred reactor, they found that the reaction in microreactor possessed significantly higher TOF value, higher and stable reaction rate, and little effect by the CO2/epoxide molar ratio. It was proposed that the activation of epoxides coordinated to the electrophilic (salen)AlCl and ring opening by the attack of nucleophilic nBu4NBr is the rate determining step, and this "electrophile–nucleophile" synergistic effect could be intensified in the microreactor, thus further improve the catalytic activity.
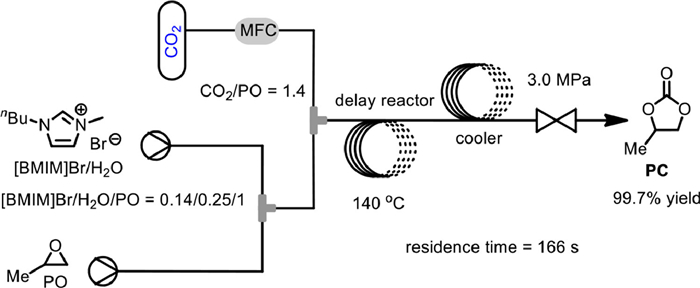
In 2021, Xu and Wang reported the synthesis of PC from PO and CO2 based on a highly efficient continuous-flow microreaction system using 1-butyl-3-methylimidazolium bromide ([BMIM]Br) as catalyst (Scheme 8) [57]. The influence of various reaction parameters was systematically studied. Finally, by setting the molar ratio of [BMIM]Br/H2O/PO to be 0.14/0.25/1 and that of CO2/PO to be 1.4, the PC could be obtained in 99.7% yield at 140 ℃ and 3.0 MPa with the residence time of 166 s. The flow pattern inside the microreaction system was identified to be varied between slug flow and annular flow. The recycling performance of [BMIM]Br catalyst was also investigated, and it could be recycled via vacuum distillation and reused for 5 times giving more than 86% yield without erosion of selectivity.
In 2021, Selva and Fiorani et al. developed a binary system consisting of NaBr/diethylene glycol (DEG) for the continuous flow synthesis of cyclic carbonates 6 from CO2 and terminal epoxides 5 in microreactors (Scheme 9) [58]. The 1/16" stainless-steel coil with a length of 500 cm and internal volume of 0.157 cm3 was employed as microfluidic reactor. The solution of epoxides 5, NaBr in DEG was delivered by HPLC pump, and CO2 was supplied as a liquid via a refrigerated dual head pump. By fixing the reaction temperature at 220 ℃, and the total pressure of 120 bar, with the flow rate of CO2 and DEG solution at 1.0 and 0.1 mL/min, respectively, a variety of cyclic carbonates 6 could be obtained in 92% to > 99% conversion with 91% to > 99% selectivity. It was proposed that DEG was not only the reaction medium, but also act as chelating agent for Na+ and hydrogen bond donor to assist the ring-opening of epoxide.
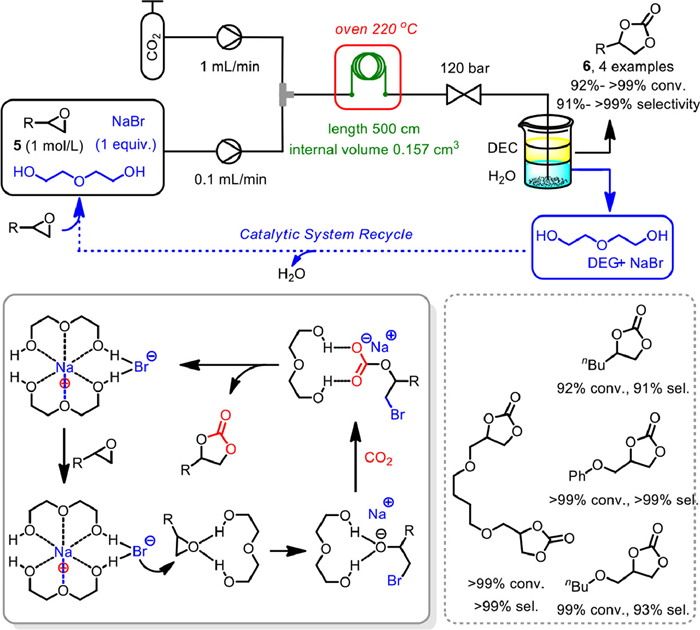
Notably, the homogeneous NaBr/DEG mixture could be recycled via a semi-continuous extraction procedure without loss of performance, which bypassed typical drawbacks associated to heterogeneous catalyst deactivation to a certain degree. The catalytic performance of NaBr/DEG binary system in batch reaction was also studied. Although the reaction conditions of batch mode were far milder, a lower productivity was obtained. The potential of continuous-flow system in terms of process intensification could also be appreciated.
As another intelligent processing of gas–liquid transformations, the application of tube-in-tube reactor to the chemical transformation of CO2 has attracted more and more attention [59]. In general, Teflon AF-2400, a chemically inert copolymer of tetrafluoroethylene and 2,2-bis(trifluoromethyl)-4,5-difluoro-1,3-dioxole, is employed as the inner gas permeable membrane tubing. As an extensively microporous, amorphous material with high gas permeability, it could effectively deliver CO2 gas to the liquid flow stream in a controlled manner, thus increase the concentration of CO2 in the system and improve the reaction efficiency.
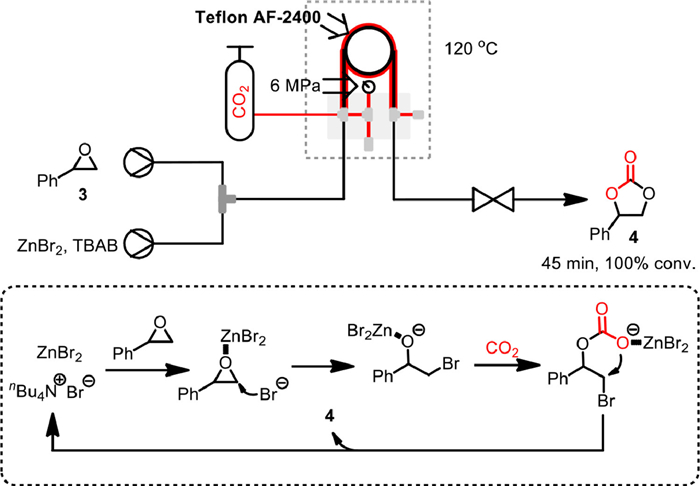
In 2018, Rehman and co-workers reported the synthesis of styrene carbonate 4 from styrene oxide 3 and CO2 in a "tube-in-tube" gas–liquid continuous flow reactor, using tetrabutylammonium bromide (TBAB) and ZnBr2 as an efficient and economical binary homogeneous catalyst (Scheme 10) [60]. The experiments were performed in a gas–liquid tube-in-tube coil reactor fits over a Uniqsis FlowSyn operating system with Teflon AF-2400 semipermeable membrane as inner tube. The rapid permeability of CO2 through the membrane, together with the high surface area to volume ratio in such continuous flow reactors, enabled the complete conversion of styrene oxide 3 within significantly reduced 45 min under 120 ℃ and 6 bar of CO2 pressure. Based on detailed kinetic study, the reaction was determined to be first-order in styrene oxide, TBAB and CO2 concentrations. Meanwhile, by using ZnBr2 as a co-catalyst, the activation energy of TBAB catalyzed reaction could be significantly reduced from 55 kJ/mol to 32 kJ/mol, clearly demonstrating the importance of synergistic effect.
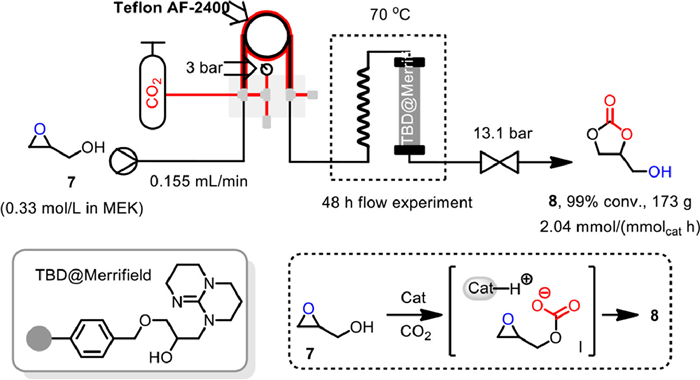
In 2021, Kleij and Pericàs reported the first continuous flow preparation of glycerol carbonate 8 from glycidol 7 and CO2 with Merrifield resin supported TBD (TBD@Merrifield) as powerful heterogeneous, metal-free and halide-free catalyst (Scheme 11) [61]. A conceptually distinct substrate-controlled approach was involved, in which the hydroxyl group of glycidol 7 activated CO2 to form a carbonic acid intermediate Ⅰ with increased nucleophilicity, then the intramolecular attack on the oxirane under basic condition providing the glycerol carbonate 8 under essentially halide-free conditions. Notably, the tube-in-tube reactor, with the inner tube made of Teflon AF-2400 as a permeable membrane, was employed to charge the CO2 into the system at low pressure. Meanwhile, a back-pressure regulator was installed after the packed bed reactor to prevent CO2 outgassing and to retain a homogeneous solution stabilizing the flow throughput. By pumping the 0.33 mol/L solution of glycidol 7 in methyl ethyl ketone (MEK) at a rate of 0.155 mL/min into the tube-in-tube reactor having a local CO2 pressure of 3 bar, while operating the back pressure regulator at 13.1 bar, 17.3 g (147 mmol) glycerol carbonate 8 could be obtained in 48 h, under 70 ℃ and a catalyst loading of 1.59 g (1.48 mmol), with an accumulated TON of 99 and a productivity of 2.04 mmol h–1 mmolcat–1 at the steady state. The flow setup was very stable during the reaction course with no loss of catalytic performance observed.
As another important route for the chemical fixation of CO2 to valuable chemicals, the direct carboxylation of carbon nucleophiles with CO2 has attracted increasing attention due to the associated high atom economy. Moreover, the resulting carboxylic acids and derivatives have wide application in pharmaceuticals, agrochemicals and advanced materials [62-64]. However, due to the inherent thermodynamical stability and kinetic inertness of CO2 gas, special strategies are required to activate CO2 or carbon nucleophiles to enable the C-C bond constructions. Consequently, the combination of flow chemistry with photocatalysis or electrosynthesis as well as the involvement of highly active organic metal intermediates for CO2 participated carboxylation reaction has been investigated and well developed in recent years.
The reaction of strong nucleophilic carbon like Grignard and organolithium serves as a powerful method for synthesizing carboxylic acids. However, the accessibility of such nucleophilic carbon, especially the highly reactive organolithium species is limited, as the latent organometallic reagents are dangerous and difficult to handle. Notably, flow chemistry techniques offer potential utility for such organometallic reagent participated reactions, as the reactive components could be safely handled in a fully contained system with the temperature controlled accurately, thus avoiding the formation of side products [65]. Moreover, it enables the in-suit generation of organometallic reagents and the subsequent carboxylation with CO2 to be performed in a more maneuverable sequence manner.
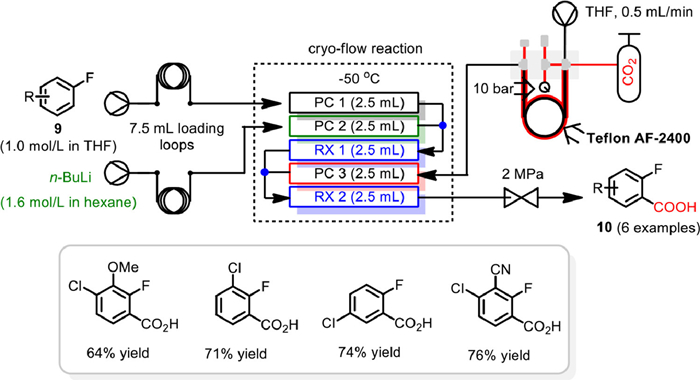
In 2014, Ley and Browne developed a continuous flow chemistry platform and applied to a broad range of low temperature organometallic reactions [66]. Based on the essence of running low temperature organometallic reactions in batch, a novel flow chemistry platform consisting of three precooling-coils (PC 1 to 3) and two reaction coils (RX 1 and 2) with independently variable volume was designed. By stacking the coils over a central cryo-cooling device, the latent organometallic reagents, the organometallic species as well as the electrophile streams could be precooled independently in the precooling-coils and then combined at the appropriate points and passed in to the appropriate reaction coils. With this new developed flow reaction system, 20-compound library of polysubstituted, fluorine-containing aromatic substrates were synthesized effectively via a sequential metalation quench procedure. Notably, the carboxylation of aryl lithium intermediate with CO2 was also realized using a fluorine-directed ortho-metalation approach (Scheme 12). By employing a Teflon AF-2400 gas permeable membrane tube-in-tube reactor to deliver CO2 as electrophile, and fixing a back pressure regulator to the output of the platform to ensure CO2 dissolving in solution, a variety of fluoroaryl carboxylic acids 10 could be obtained in up to 76% yield from simple fluorobenzenes 9. These results illustrated this system might be operated as a supportive device to provide early scale-up and deliver quantities of material suitable for application testing.
In 2019, an efficient and atom-economic three-step flow synthesis of ibuprofen from p-xylene was realized by Kim and co-workers, based on a sequential benzylic C-H metalations manner (Scheme 13) [67]. The first two steps alkylation were realized in a capillary microreactor consisting of two micromixers M1 and M2 and two tube reactors R1 and R2, with the LICKOR type superbase generated in situ from tBuLi and tBuOK for the selective benzylic C-H metalation. The mixture of starting material with tBuOK was mixed with tBuLi in M1 and reacted in R1, then the resulting solution was sequentially mixed with alkylation reagent in M2 and reacted in R2. By varying the concentration, solvent, base equivalents, temperature and precisely controlled the residence time via changing the length of tube reactor, the methylation and isopropylation product 11 and 12 was obtained in 95% and 93% yield with MeOTf and isopropyl iodide as alkylation reagent, respectively.
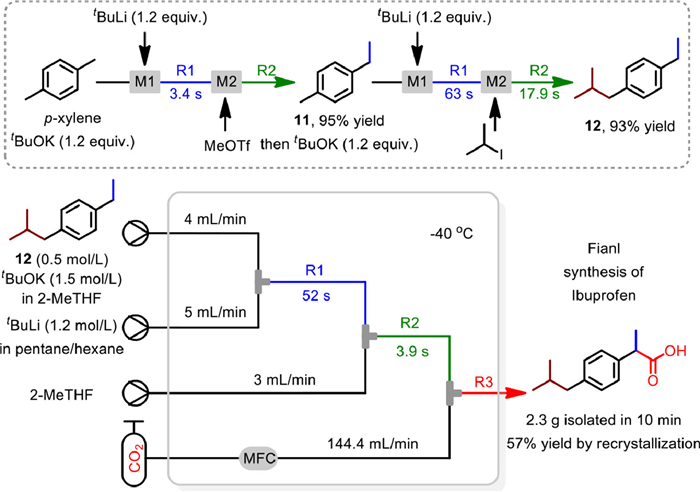
The final synthesis of ibuprofen was conducted by biphasic reaction with CO2 as electrophile, using a flow microreactor constructing with three micromixers and three tube reactors (R1 to R3). Considering the difficultly in metalation of the benzyl position of 12, caused by the steric hindrance and electronic nature, 3.0 equiv. of tBuLi/tBuOK was employed with a longer 52 s residence time in R1. Then, the resulting solution was slightly diluted with 2-MeTHF, to benefit the dissolution of CO2 and avoid the possible clogging issue, with 3.9 s of residence time in R2. Finally, the introduction of three equivalents of CO2 gave ibuprofen effectively via a biphasic segment flow. Notably, by continuously operating the flow reaction for 10 min, 2.3 g of ibuprofen could be obtained with 57% isolated yield by recrystallization. The gram-scale productivity of ibuprofen as well as the effective inhibition of by-products, clearly demonstrated the superiority and practicability of flow chemistry.
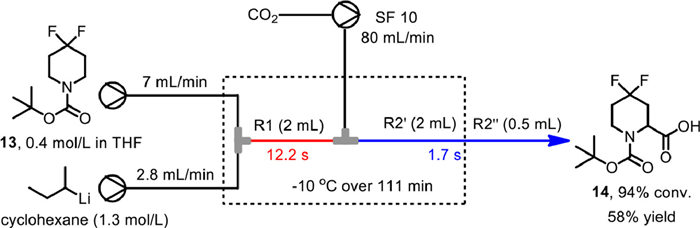
In 2022, Matthieu and co-workers reported the large-scale carboxylation of N-Boc-4,4-difluoropiperidine 13 based on continuous flow process (Scheme 14) [68]. At first a Vapourtec setup was used for the initial condition optimization, in which a Uniqsis glass microreactor R1 was used for the N-Boc-directed α-deprotonation of s-BuLi, and a coil PTFE reactor R2 (R2' + R2'') was used for the subsequent trapping of the lithiated piperidine with CO2. Meanwhile, CO2 was dispensed by a Vapourtec SF10 peristaltic pump. By fixing the residence times in R1 and R2 with 12.2 and 1.7 s, and adjusting the flow rates of s-BuLi, difluoropiperidine 13 and CO2 in 2.8, 7 and 80 mL/min, the flow reaction under -10 ℃ over 111 min without interruption could transform 69 g of difluoropiperidine into carboxylic acid in 58% isolated yield with 94% conversion.
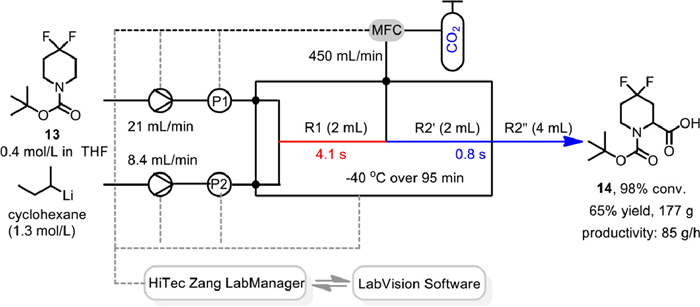
Then, to get a larger quantity and improve safety, a single glass microreactor consisting of two sections (R1 = 2 mL and R2' = 2 mL) and equipping with integrated heat exchangers connected to a thermostat was employed for the lithiation/carboxylation sequence (Scheme 15). All the devices, including Syrdos syringe pumps to deliver s-BuLi and difluoropiperidine, MFC for CO2, thermostat and pressure sensors, were connected to a HiTec Zang LabManager and controlled by LabVision software. By increasing the flow rates of s-BuLi, difluoropiperidine 13 and CO2 to 8.4, 21 and 450 mL/min, with residence times in R1 and R2 (R2' + R2'') shortened to 4.1 and 0.8 s, respectively, the scale-up flow experiment under -40 ℃ over 95 min with no interruption, could deliver the desired carboxylic acid 14 in 65% overall yield with a productivity of 85 g/h. As the carboxylic acid lithium salt precipitation at the mixing point led to clogging and an increase in pressure in the system, a washing procedure was needed during the production campaign between each run. Finally, 400 g of carboxylic acid could be obtained over the course of a day based on the established safe and scalable flow process. This process clearly demonstrated the powerfulness of flow chemistry in the organolithium mediated carboxylation reaction.
In principle, the chemical fixation CO2 processes could be accelerated by accessing radical mechanisms involving single-electron species as a strategy to overcome the inherent barriers [69]. The employment of photocatalysis has been important to the discovery of novel carboxylation reactions, as it could convert light energy to chemical energy by harvesting photons and subsequently generating reactive intermediates via single electron transfer manner [70-74]. Moreover, with the combination of flow chemistry, the increased surface area during the photocatalysis enables more efficient irradiation of photocatalyst, thus leading to shorter reaction times and facile scale-up [75].
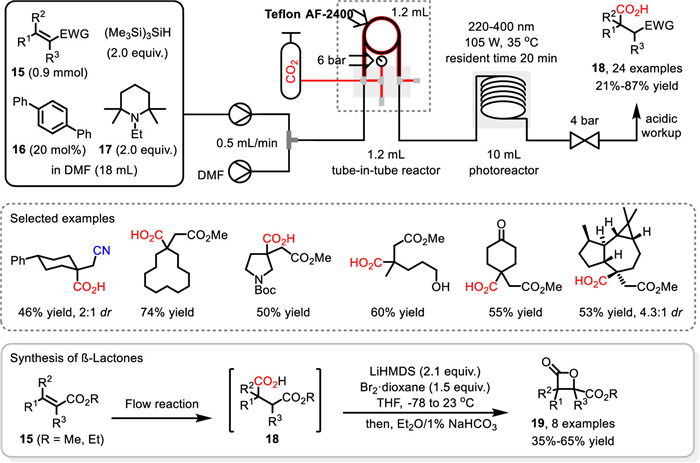
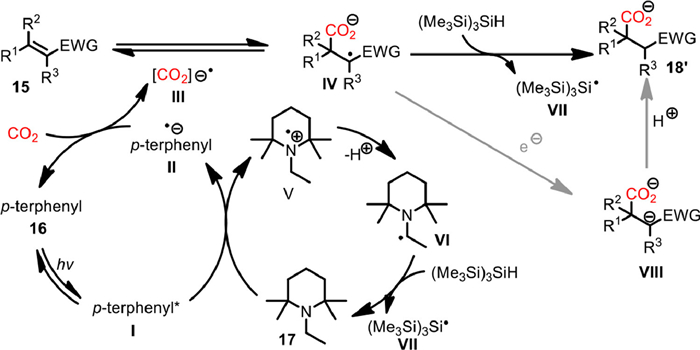
In 2021, Romo and co-workers realized the first photocatalyzed β-selective hydrocarboxylation of α,β-unsaturated ester, nitriles and ketones with CO2 under flow conditions (Scheme 16) [76]. The pivotal UV excitation was conducted in 10 mL photochemical flow reactor with wavelength of 220-400 nm (105 W). Meanwhile, the tube-in-tube reactor was used to ensure the efficient delivery of CO2 (6 bar) to the liquid flow stream. With p-terphenyl 16 as photocatalyst and N-ethyl-2,2,6,6-tetramethylpiperidine 17 as electron donor as well as bulky (TMS)3SiH as hydrogen atom donor, the photoexcitation at 35 ℃ with a total resident time of 20 min enabled the regioselective flow hydrocarboxylation of various of α,β-unsaturated ester, nitriles and ketones 15 to proceed smoothly to deliver the corresponding carboxylic acids 18 in up to 87% yield. Some of the carboxylic acids bearing all-carbon quaternary centers could also be accessed in good to excellent yield. Furthermore, with the thus developed β-selective flow hydrocarboxylation as key step, the telescoped synthesis of β-lactones 19 was also realized from α,β-unsaturated esters 15, through a subsequent α-bromination and β-lactonization sequence. A variety of bicyclic and spiro β-lactones could be obtained effectively without purification of intermediates.
Based on systematically investigation and previous reports, a possible reaction mechanism for the photocatalyzed β-selective hydrocarboxylation was proposed (Scheme 17). Initially, the photoexcitation of p-terphenyl 16 gave excited singlet state of p-terphenyl* Ⅰ. The subsequent single electron transfer with amine 17 delivered the reduced p-terphenyl radical anion Ⅱ, which could reduce CO2 to CO2 radical anion Ⅲ, thus initiating a Giese-type addition to the α,β-unsaturated ester, nitriles or ketones 15. Finally, the quenching of the generated radical anion Ⅳ with a hydrogen atom from (TMS)3SiH gave the final carboxylate product 18'. The further reduction of radical anion Ⅳ and following protonation of Ⅷ might also be involved during the reaction course.
Apart from photocatalysis, electrosynthesis represents another useful tool for the chemical fixation of CO2, as it could overcome the inherent thermodynamic stability of CO2 by using electrons as redox reagents [77-84]. Moreover, it affords a very facile and precise way to generate highly energetic intermediates via control of the electrode potential, providing good alternatives to traditional chemical methods. In this context, electrocarboxylation has the potential to address sustainability from not only a raw materials perspective but also an energetic perspective. In this vein, the combination of electrosynthesis with flow chemistry has been a promising general strategy for carboxylation reactions of CO2 [85,86].
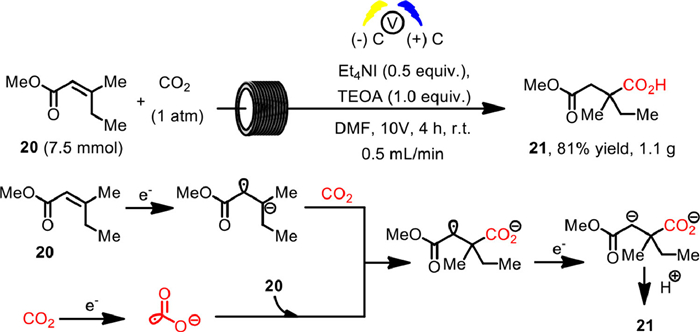
In 2021, Buckley and co-workers developed the first highly selective electrochemical hydrocarboxylation of α,β-unsaturated esters with CO2 [87]. Using triethanolamine (TEOA) as a proton source, Et4NI as electrolyte, the constant voltage electrolysis with inert carbon electrode in DMF enabled the β-carboxylation to give all-carbon α-quaternary centered carboxylic acids in good yield. The corresponding electrochemical flow process was also conducted in a Vapourtech ion electrochemical reactor fixing carbon–carbon sheet electrodes (Scheme 18). The reaction of α,β-unsaturated ester 20 with CO2 at a constant voltage of 10 V in closed cycle with speed of 0.5 mL/min for 4 h gave the desired product 21 in 81% yield on a gram scale, which could be isolated by simple crystallization, further demonstrating the simplicity of their approach. It was proposed that the initial reduction of α,β-unsaturated ester 20 or CO2 were all possible routes to the final mono-carboxylated product 21. Meanwhile, the high regioselectivity could be explained by the DFT calculation of Lan and Yu [88], where the acrylate radical anion complex possessed higher spin density in the β-position.
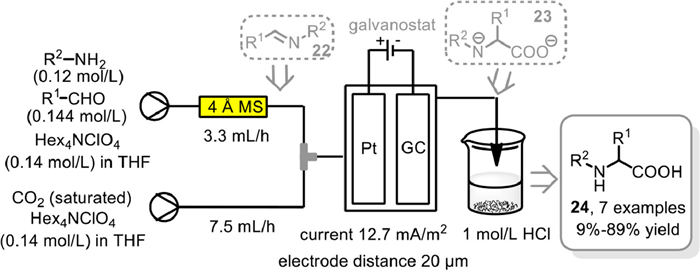
The α-carboxylation of amine derivatives with CO2 represents to be as a reasonable and economical route to α-amino acids in particular the unnatural ones, with the carboxyl unit derived from CO2 directly. As early as 2017, Atobe and co-workers reported a direct electrochemical carboxylation of aldimines in a flow microreactor for the synthesis of α-amino acids [89]. However, the instability of aldimines restricted the application of this method to some extent. Therefore, an integrated flow synthetic system was developed in 2021 by the same group, in which the aldimines was generated in-situ and applied directly to the electrochemical carboxylation in a flow operation manner (Scheme 19) [90]. A desiccant bed column with 20 cm long and 1.0 cm inner diameter filled with 4 Å MS as desiccant was employed for the initial dehydration–condensation. Meanwhile, the subsequent electrochemical carboxylation of the thus in-suit formed aldimine 22 with CO2 was conducted in electrochemical flow microreactor with Hex4NClO4 electrolyte in THF using constant-current mode at current density of 12.7 mA/cm2. Moreover, the rapid acidification could avoid the decomposition of thus formed unstable carboxylate ions, thus enabled the electrochemical carboxylation to be performed in the absence of sacrificial anode. Finally, not only aryl-substituted aldimine intermediates, but also more demanding alkyl-substituted ones were all tolerated to deliver the corresponding α-amino acids 24 in up to 89% yields. These results clearly demonstrated the unique advantages of flow chemistry in multistep synthesis progress.
In the last four years, significant advancements have been made in flow chemistry enabled reactions using CO2 as a C1 synthon, which have been summarized in this review. In particular, packed-bed type, coil type and chip type reactor as well as the tube in tube reactor have all been applied to the chemical fixation of CO2. Examples of heterogeneous catalysis in packed-bed reactor and homogeneous catalysis in microreactors provide efficient routes for the coupling of CO2 with epoxides to cyclic carbonates with shorter reaction times, lower catalyst loadings, and better sustainability. Based on flow technology enabled C-H metalation, a series of carboxylic acids and their derivatives have been constructed from CO2 and simple aromatics and alkanes. Moreover, electrosynthesis and photocatalysis has also been employed for the carboxylation of CO2 in continuous flow systems, based on the facts that electrochemistry and photochemistry could generate highly energetic intermediates in a facile and precise way.
Despite these tremendous achievements, there is still ample room for chemical fixation of CO2 under continuous flow systems to valuable chemicals. First of all, more successful examples are needed to demonstrate the feasibility and potential of continuous-flow techniques, based on the fact that there have been more and more synthetically important reactions developed using CO2 as C1 synthon. Secondary, multistep continuous-flow synthesis involving CO2 as carboxylation reagent is emerging and successful examples have been shown here. Although these results represent a major leap toward the real-world active pharmaceutical ingredients synthesis, the scope of such elegant transformations needs further demonstration. Thirdly, apart from electrosynthesis and photocatalysis, enzymatic catalysis also represents a very promising approach for continuous-flow manufacturing [91], more and more successful examples are expected. Finally, considering the rapid and exciting progress in CO2 participated asymmetric synthesis [92-95], successful examples of continuous-flow asymmetric catalysis for the chemical fixation of CO2 are foreseeable.
In light of advantages of continuous flow systems, it is hoped that with the further development of new catalytic systems, new synthetic strategy and new engineering techniques, more and more attractive and useful CO2-based continuous-flow transformations will be exploited. Considering the ever-growing number of contributions to this very promising but also challenging research field, a bright future can be forecast.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The financial support from the National Key Research and Development Program of China (No. 2020YFA0710200), National Natural Science Foundation of China (No. 22171090), the Shanghai Science and Technology Innovation Action Plan (No. 20JC1416900) and the Fundamental Research Funds for the Central Universities is highly appreciated. We thank Prof. Wei-Ping Zhu and Li Zhang at East China University of Science and Technology for the guidance on flow chemistry.
N. Mac Dowell, P. S. Fennell, N. Shah, G. C. Maitland, Nat. Clim. Change 7 (2017) 243–249. doi: 10.1038/nclimate3231
K. B. Tokarska, K. Zickfeld, Environ. Res. Lett. 2015, 10, 094013 doi: 10.1088/1748-9326/10/9/094013
E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas, C. W. Jones, Chem. Rev. 116 (2016) 11840–11876. doi: 10.1021/acs.chemrev.6b00173
B. Walsh, P. Ciais, I. A. Janssens, et al., Nat. Commun. 8 (2017) 14856. doi: 10.1038/ncomms14856
M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010.
B.M. Bhanage, M. Arai, Transformation and Utilization of Carbon Dioxide, Springer, Berlin, Heidelberg, 2014.
D. M. D'Alessandro, B. Smit, J. R. Long, Angew. Chem. Int. Ed. 49 (2010) 6058–6082. doi: 10.1002/anie.201000431
M. Aresta, A. Dibenedetto, A. Angelini, Chem. Rev. 114 (2014) 1709–1742. doi: 10.1021/cr4002758
A. D. Tjandra, J. Huang, Chin. Chem. Lett. 29 (2018) 734–746. doi: 10.1016/j.cclet.2018.03.017
Lu, X. -B. Top. Organomet. Chem. 53 (2015) 171–198.
M. M. F. Hasan, L. M. Rossi, D. P. Debecker, et al., ACS Sustain. Chem. Eng. 9 (2021) 12427–12430. doi: 10.1021/acssuschemeng.1c06008
Z. Gao, J. Li, Z. Zhang, W. Hu, Chin. Chem. Lett. 33 (2022) 2270–2280. doi: 10.1016/j.cclet.2021.09.037
M. He, Y. Sun, B. Han, Angew. Chem. Int. Ed. 52 (2013) 9620–9630. doi: 10.1002/anie.201209384
M. He, Y. Sun, B. Han, Angew. Chem. Int. Ed. 61 (2022) e202112835.
X.B. Lu, Carbon Dioxide and Organometallics, Springer, Cham, Switzerland, 2016.
M. Aresta, A. Dibenedetto, E. Quaranta, Reaction Mechanisms in Carbon Dioxide Conversion, Springer, Berlin, Heidelberg, 2016.
Q. Liu, L. Wu, R. Jackstell, M. Beller, Nat. Commun. 6 (2015) 5933–5948. doi: 10.1038/ncomms6933
J. Artz, T. E. Müller, K. Thenert, et al., Chem. Rev. 118 (2018) 434-504. doi: 10.1021/acs.chemrev.7b00435
S. Dabral, T. Schaub, Adv. Synth. Catal. 361 (2019) 223–246. doi: 10.1002/adsc.201801215
M. B. Plutschack, B. Pieber, K. Gilmore, P. H. Seeberger, Chem. Rev. 117 (2017) 11796–11893. doi: 10.1021/acs.chemrev.7b00183
B. Endrődi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar, C. Janáky, Prog. Energy Combust. Sci. 62 (2017) 133–154. doi: 10.1016/j.pecs.2017.05.005
F. M. Akwi, P. Watts, Chem. Commun. 54 (2018) 13894–13928. doi: 10.1039/c8cc07427e
M. Guidi, P. H. Seeberger, K. Gilmore, Chem. Soc. Rev. 49 (2020) 8910–8932. doi: 10.1039/c9cs00832b
M. Trojanowicz, Molecules 25 (2020) 1434. doi: 10.3390/molecules25061434
J. Xie, D. Zhao, Chin. Chem. Lett. 31 (2020) 2395–2400. doi: 10.1016/j.cclet.2020.03.022
C. J. Mallia, I. R. Baxendale, Org. Process Res. Dev. 20 (2016) 327–360. doi: 10.1021/acs.oprd.5b00222
J. A.M. Lummiss, P. D. Morse, R. L. Beingessner, T. F. Jamison, Chem. Rec. 17 (2017) 667–680. doi: 10.1002/tcr.201600139
P. D. Morse, R. L. Beingessner, T. F. Jamison, Isr. J. Chem. 57 (2017) 218–227. doi: 10.1002/ijch.201600095
J. Wegner, S. Ceylan, A. Kirschning, Adv. Synth. Catal. 354 (2012) 17–57. doi: 10.1002/adsc.201100584
B. Gutmann, D. Cantillo, C. O. Kappe, Angew. Chem. Int. Ed. 54 (2015) 6688–6728. doi: 10.1002/anie.201409318
M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, et al., Chem. Soc. Rev. 45 (2016) 4892–4928. doi: 10.1039/C5CS00902B
I. Rossetti, M. Compagnoni, Chem. Eng. J. 296 (2016) 56–70. doi: 10.1016/j.cej.2016.02.119
T. Sakakura, J.C. Choi, H. Yasuda, Chem. Rev. 107 (2007) 2365–2387. doi: 10.1021/cr068357u
C. Das Neves Gomes, O. Jacquet, C. Villiers, et al., Angew. Chem. Int. Ed. 51 (2012) 187–190. doi: 10.1002/anie.201105516
Q.W. Song, Z.H. Zhou, L.N. He, Green Chem. 19 (2017) 3707–3728. doi: 10.1039/C7GC00199A
A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas, R. Martin, Angew. Chem. Int. Ed. 57 (2018) 15948–15982. doi: 10.1002/anie.201803186
C. Zhou, M. Li, J. Yu, S. Sun, J. Cheng, Chin. J. Org. Chem. 40 (2020) 2221–2231. doi: 10.6023/cjoc202003039
J. Liu, C. Chen, K. Zhang, L. Zhang, Chin. Chem. Lett. 32 (2021) 649–659. doi: 10.1016/j.cclet.2020.07.040
Y. Yi, W. Hang, C. Xi, Chin. J. Org. Chem. 41 (2021) 80–93. doi: 10.6023/cjoc202007013
J.H. Ye, T. Ju, H. Huang, L.L. Liao, D.G. Yu, Acc. Chem. Res. 54 (2021) 2518–2531. doi: 10.1021/acs.accounts.1c00135
C.K. Ran, L.L. Liao, T.Y. Gao, Y.Y. Gui, D.G. Yu, Curr. Opin. Green Sustain. Chem. 32 (2021) 100525. doi: 10.1016/j.cogsc.2021.100525
H. Seo, L. V. Nguyen, T. F. Jamison, Adv. Synth. Catal. 361 (2019) 247–264. doi: 10.1002/adsc.201801228
M. North, R. Pasquale, C. Young, Green Chem. 12 (2010) 1514–1539. doi: 10.1039/c0gc00065e
C. Martín, G. Fiorani, A. W. Kleij, ACS Catal. 5 (2015) 1353–1370. doi: 10.1021/cs5018997
B. Schäffner, F. Schäffner, S. P. Verevkin, A. Börner, Chem. Rev. 110 (2010) 4554–4581. doi: 10.1021/cr900393d
M. Lichtenwalter, J. Cooper, US Patent, 2873282, 1959.
P. P. Pescarmona, M. Taherimehr, Catal. Sci. Technol. 2 (2012) 2169–2187. doi: 10.1039/c2cy20365k
T. Takahashi, T. Watahiki, S. Kitazume, H. Yasuda, T. Sakakura, Chem. Commun. (2006) 1664–1666. doi: 10.1039/b517140g
Y. Zhang, Z. Tan, B. Liu, D. Mao, C. Xiong, Catal. Commun. 68 (2015) 73–76. doi: 10.1016/j.catcom.2015.05.004
T. Wang, W. Wang, Y. Lyu, et al., RSC Adv. 7 (2017) 2836–2841. doi: 10.1039/C6RA26780G
D. Valverde, R. Porcar, P. Lozano, E. García-Verdugo, S. V. Luis, ACS Sustain. Chem. Eng. 9 (2021) 2309–2318. doi: 10.1021/acssuschemeng.0c08388
J. Sun, Z. Li, J. Yin, J. CO2 Util. 53 (2021) 101723. doi: 10.1016/j.jcou.2021.101723
T. Q. Bui, L. J. Konwar, A. Samikannu, D. Nikjoo, J.P. Mikkola, ACS Sustain. Chem. Eng. 8 (2020) 12852–12869. doi: 10.1021/acssuschemeng.0c03123
P. T. Baraldi, V. Hessel, Green Process. Synth. 1 (2012) 149–167.
J. Deng, J. Zhang, K. Wang, G. Luo, Chin. J. Chem. 37 (2019) 161–170. doi: 10.1002/cjoc.201800428
M.R. Li, M.C. Zhang, T.J. Yue, X.B. Lu, W.M. Ren, RSC Adv. 8 (2018) 39182–39186. doi: 10.1039/c8ra07236a
Y. Wu, Y. Ding, J. Xu, et al., Green Energy Environ. 6 (2021) 291–297. doi: 10.1016/j.gee.2020.04.016
D. Rigo, R. Calmanti, A. Perosa, M. Selva, G. Fiorani, ChemCatChem 13 (2021) 2005–2016. doi: 10.1002/cctc.202002010
M. Brzozowski, M. O'Brien, S. V. Ley, A. Polyzos, Acc. Chem. Res. 48 (2015) 349–362. doi: 10.1021/ar500359m
A. Rehman, A. M. L. Fernández, M.F.M. G. Resul, A. Harvey, J. CO2 Util. 24 (2018) 341–349. doi: 10.1016/j.jcou.2018.02.001
N. Zanda, A. Sobolewska, E. Alza, A. W. Kleij, M. A. Pericas̀, ACS Sustain. Chem. Eng. 9 (2021) 4391–4397. doi: 10.1021/acssuschemeng.1c01060
X.F. Wu, F. Zheng, Top. Curr. Chem. 375 (2017) 4. doi: 10.1007/s41061-016-0091-6
S.S. Yan, Q. Fu, L.L. Liao, et al., Coord. Chem. Rev. 374 (2018) 439–463. doi: 10.1016/j.ccr.2018.07.011
M. Börjesson, T. Moragas, D. Gallego, R. Martin, ACS Catal. 6 (2016) 6739–6749. doi: 10.1021/acscatal.6b02124
M. Power, E. Alcock, G. P. McGlacken, Org. Process Res. Dev. 24 (2020) 1814–1838. doi: 10.1021/acs.oprd.0c00090
J. A. Newby, W. Blaylock, P. M. Witt, et al., Org. Process Res. Dev. 18 (2014) 1211–1220. doi: 10.1021/op500213j
H.J. Lee, H. Kim, D.P. Kim, Chem. Eur. J. 25 (2019) 11641–11645. doi: 10.1002/chem.201903267
J.P. Kestemont, J. R. Frost, J. Jacq, et al., Org. Process Res. Dev. 26 (2022) 635–639. doi: 10.1021/acs.oprd.1c00092
Y. Yang, Y. Tang, H. Jiang, et al., Chin. Chem. Lett. 30 (2019) 2089–2109. doi: 10.1016/j.cclet.2019.10.041
C. S. Yeung, Angew. Chem. Int. Ed. 58 (2019) 5492–5502. doi: 10.1002/anie.201806285
Z. Zhang, J. Ye, T. Ju, et al., ACS Catal. 10 (2020) 10871–10885. doi: 10.1021/acscatal.0c03127
B. Cai, H. W. Cheo, T. Liu, J. Wu, Angew. Chem. Int. Ed. 60 (2021) 18950–18980. doi: 10.1002/anie.202010710
Z. Fan, Z. Zhang, C. Xi, ChemSusChem 13 (2020) 6201–6218.
X. He, L.Q. Qiu, W.J. Wang, K.H. Chen, L.N. He, Green Chem. 22 (2020) 7301–7320. doi: 10.1039/d0gc02743j
L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag, T. Noël, Chem. Rev. 122 (2022) 2752–2906. doi: 10.1021/acs.chemrev.1c00332
G. Kang, D. Romo, ACS Catal. 11 (2021) 1309–1315. doi: 10.1021/acscatal.0c05050
X.F. Liu, K. Zhang, L. Tao, X.B. Lu, W.Z. Zhang, Green Chem. Eng. 3 (2022) 125–137. doi: 10.3390/catal12020125
P. D. Luna, C. Hahn, D. Higgins, et al., Science 364 (2019) 350.
Y. Cao, X. He, N. Wang, H.R. Li, L.N. He, Chin. J. Chem. 36 (2018) 644–659. doi: 10.1002/cjoc.201700742
A. Cherubini-Celli, J. Mateos, M. Bonchio, L. Dell'Amico, X. Companyó, ChemSusChem 11 (2018) 3056–3070. doi: 10.1002/cssc.201801063
H. Senboku, A. Katayama, Curr. Opin. Green Sustain. Chem. 3 (2017) 50–54. doi: 10.1016/j.cogsc.2016.10.003
L. Rossi, Curr. Green Chem. 2 (2015) 77–89. doi: 10.2174/2213346101666140804222344
R. Matthessen, J. Fransaer, K. Binnemans, D. E. De Vos, Beilstein J. Org. Chem. 10 (2014) 2484–2500. doi: 10.3762/bjoc.10.260
B.P. Sullivan, K. Krist, H.E. Guard, Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, Elsevier, Amsterdam, 1993.
D. Pletcher, R. A. Green, R. C. D. Brown, Chem. Rev. 118 (2018) 4573–4591. doi: 10.1021/acs.chemrev.7b00360
T. Noël, Y. Cao, G. Laudadio, Acc. Chem. Res. 52 (2019) 2858–2869. doi: 10.1021/acs.accounts.9b00412
A. M. Sheta, A. Alkayal, M. A. Mashaly, et al., Angew. Chem. Int. Ed. 60 (2021) 21832–21837. doi: 10.1002/anie.202105490
H. Huang, J.H. Ye, L. Zhu, et al., CCS Chem. 2 (2020) 1746–1756.
Y. Qu, C. Tsuneishi, H. Tateno, Y. Matsumura, M. Atobe, React. Chem. Eng. 2 (2017) 871–875. doi: 10.1039/C7RE00149E
Y. Naito, Y. Nakamura, N. Shida, et al., J. Org. Chem. 86 (2021) 15953–15960. doi: 10.1021/acs.joc.1c00821
T. Matsuda, R. Marukado, S. Koguchi, et al., Tetrahedron Lett. 49 (2008) 6019–6020. doi: 10.1016/j.tetlet.2008.08.004
J. Vaitla, Y. Guttormsen, J. K. Mannisto, et al., ACS Catal. 7 (2017) 7231–7244. doi: 10.1021/acscatal.7b02306
X. Guo, Y. Wang, J. Chen, G. Li, J.B. Xia, Chin. J. Org. Chem. 40 (2020) 2208–2220. doi: 10.6023/cjoc202002032
C.K. Ran, X.W. Chen, Y.Y. Gui, et al., Sci. China Chem. 63 (2020) 1336–1351. doi: 10.1007/s11426-020-9788-2
Y. Shi, B.W. Pan, Y. Zhou, et al., Org. Biomol. Chem. 18 (2020) 8597–8619. doi: 10.1039/d0ob01905d

Figure 1 Schematic diagram of reaction device. Reproduced with permission [42]. Copyright 2019, Wiley-VCH.

Scheme 1 SiO2-C3H6-P(n-Bu)3Br catalyzed coupling of PO with CO2. Reproduced with permission [48]. Copyright 2006, Royal Society of Chemistry.

Scheme 2 CSAC tethered ionic liquids for coupling of epoxide with CO2. Reproduced with permission [49]. Copyright 2015, Elsevier.

Scheme 3 Polymer supported ionic liquid catalyzed coupling of PO with CO2. Reproduced with permission [50]. Copyright 2017, Royal Society of Chemistry.

Scheme 4 Rose Bengal based polymer catalyzed coupling reaction. Reproduced with permission [51]. Copyright 2021, American Chemical Society.

Scheme 5 DBU-based ionic liquid catalyzed coupling of PO with CO2. Reproduced with permission [52]. Copyright 2021, Elsevier.

Scheme 6 MFFR catalyzed coupling of epoxide with CO2. Reproduced with permission [53]. Copyright 2020, American Chemical Society.

Scheme 7 (Salen) AlCl catalyzed coupling reaction of CO2 in microreactor. Reproduced with permission [56]. Copyright 2018, Royal Society of Chemistry.

Scheme 8 [BMIM]Br catalyzed coupling of PO with CO2. Reproduced with permission [57]. Copyright 2021, Chinese Academy of Sciences.

Scheme 9 Diethylene glycol/NaBr catalyzed coupling of PO with CO2. Reproduced with permission [58]. Copyright 2021, Wiley-VCH.

Scheme 10 ZnBr2/TBAB catalyzed reaction in tube-in-tube reactor. Reproduced with permission [60]. Copyright 2018, Elsevier.

Scheme 11 TBD@Merrifield catalyzed reaction in tube-in-tube reactor. Reproduced with permission [61]. Copyright 2021, American Chemical Society.

Scheme 12 Low-temperature flow platform enabled carboxylation. Reproduced with permission [66]. Copyright 2014, American Chemical Society.

Scheme 13 Three-step synthesis of ibuprofen from p-xylene. Reproduced with permission [67]. Copyright 2019, Wiley-VCH.

Scheme 14 Carboxylation of 4,4-difluoropiperidine on Vapourtec setup. Reproduced with permission [68]. Copyright 2022, American Chemical Society.

Scheme 15 Scale-up of flow carboxylation of N-Boc-4,4-difluoropiperidine. Reproduced with permission [68]. Copyright 2022, American Chemical Society.

Scheme 16 Photocatalyzed flow hydrocarboxylation. Reproduced with permission [76]. Copyright 2021, American Chemical Society.

Scheme 17 Proposed mechanism and telescoped synthesis of β-lactones. Reproduced with permission [76]. Copyright 2021, American Chemical Society.

Scheme 18 Electrosynthetic flow hydrocarboxylation. Reproduced with permission [87]. Copyright 2021, Wiley-VCH.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
