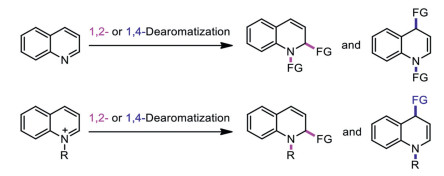
Scheme 1.
Weakened the aromaticity of pyridine rings moiety.
Metal-free nucleophilic 7, 8-dearomatization of quinolines: Spiroannulation of aminoquinoline protected amino acids
Zhiguo Zhang , Xiyang Cao , Xiaoqing Song , Gang Wang , Bingbing Shi , Xiang Li , Nana Ma , Lantao Liu , Guisheng Zhang

Arene dearomatization is an important transformation, which can easy access alicyclic frameworks existing in biologically and pharmacologically active compounds from the ubiquitous precursor. Among extant reports, most focus on dearomatization of substituted benzene compounds and pyridine derivatives [1-17]. In comparison, dearomatization of other arenes such as quinolines are far from being developed [18]. Furthermore, dearomatization of quinolines mainly occurred via 1, 2- or 1, 4-dearomatization of the pyridine moiety, whose aromaticity can be weakened for nucleophilic addition. Currently, examples of this kind include alkenylation [19, 20], acylation [21, 22], hydroboration [23–25], hydrosilylation [26, 27], allylic alkylation [28, 29], hydroamination [30], phosphonylation [31], hydrogenation [32], and other organocatalyzed cascade multiple functionalizations [33]. In addition, quinoline N-oxides and quinolinium imides were also used as the dearomatizable precursors to construct N-heterocyclic compounds [3439], as the direct functionalization of quinolines and their derivatives remains limited because of their low reactivity and poor intrinsic regioselectivity (Scheme 1).
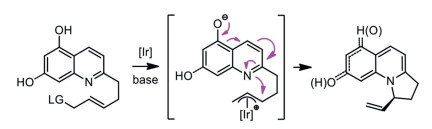
In fact, control of regioselectivity in the dearomatization of activated N-aromatic compounds has mainly relied on the electron density of the nucleophiles, namely, their hardness or softness [40]. However, no matter what nucleophiles were used, dearomatization always occurred on the pyridine moiety of quinolines. To solve these problems, hydroxyl group was pre-introduced to the benzene moiety, which may enhance the ability of the dearomatization of the benzene group and facilitate the subsequent dearomatization reaction of quinolones [41]. The procedure similar as the dearomatization of phenols [42]. It is worth noting that, in 2018, You's group realized an iridium-catalyzed intramolecular asymmetric allylic alkylation reactions starting from 5- and 7-hydroxyquinoline derivatives via 1, 5- and 1, 7-dearomatization (Scheme 2) [41]. They simultaneously weakened the aromaticity of both rings with the assistant of the hydroxyl group. Despite considerable efforts, examples of stereoselective 7, 8-dearomative functionalization have not been reported, because they are particularly less favorable in terms of steric and electric effect than dearomative reactions on the pyridine ring. Described herein is the example of metal-free nucleophilic 7, 8-dearomatization of quinolines, a highly selective weakening of the aromaticity of benzene moiety in quinolones.
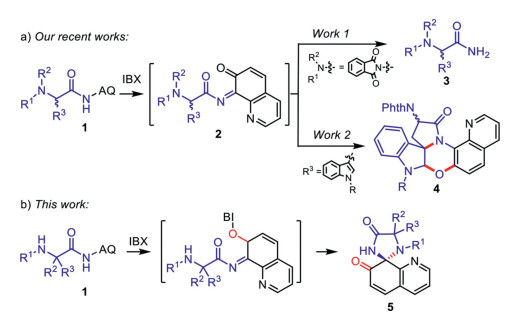
Over the past decades, synthetic organic chemistry benefited from the development of a large variety of hypervalent iodine reagents, which are valuable synthetic tools for a vast and constantly growing array of different applications [43-51]. Very recently, we reported a systematic study on the derivation reaction of N-aryl amides in the presence of polyvalent iodine(III) and iodine(V) compounds [52–56]. Among them, we reported a series of efficient and chemoselective methods to convert various secondary N-arylamides 1 to primary amides 3 in high yields by the treatment of organoiodine in mixed solvents of H2O and HFIP (Scheme 3a) [53, 55–57]. These regiospecific C(aryl)-N bond cleavage reaction without touching the C(carbonyl)-N bond in the amides, not only further enriches the amino group protecting chemistry, but also provides a way for the facile removal of the aminoquinolines (AQ) directing group under mild conditions [58–61]. As part of our ongoing program on exploring amides derivation reactions [62], our group disclosed a 2-iodoxybenzoic acid (IBX)-mediated intramolecular oxidative spiro-fused tandem cyclization reaction of tryptophan analogs 1 bearing an N-arylamides side-chain to rapidly afford polycyclic spiroindolines 4 under mild conditions [52]. Mechanistic investigation in these studies indicated that all transformations features a common imide intermediate 2 via a 7, 8-dearomatization of the aniline moiety triggered by oxidation at the ortho-position of AQ group (Scheme 3a). Prompted by these discoveries, we envisioned that intermediate 2 may be attacked by a nucleophilic reagent on the side chain such as a secondary amine, to give a spiro product 5 (Scheme 3b).
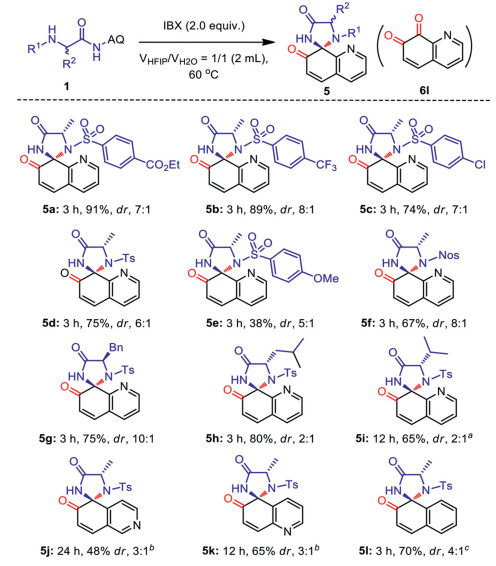
As shown in Table 1, after many attempts, we established that α-amino amide (α-AA) 1d, derived from Ala, worked well and afforded the desired spiro product 5d in 75% yield with moderate diastereoselectivity (dr > 6:1) in the presence of 2.0 equiv. of IBX in mixed solvents of HFIP and H2O (1:1) at 60 ℃ for 3 h (entry 1). The structure of 5d was confirmed by X-ray crystallography (CCDC: 2161853). The reaction carried out in the mixed solvents of HFIP and H2O in ratios of 3:1 and 1:3 both gave a slightly lower yield (entries 2 and 3). As expected, the reaction did not occur in neat H2O, with a large amount of substrate 1d being recovered because of the poor solubility of starting material and IBX (entry 4). No product was generated in the reaction performed in HFIP either (entry 5). These observations indicate that increasing solubility of the substrates is beneficial to improve the yield, while H2O played an important role in the transformation. Other fluorine-containing solvents, such as 2, 2, 3, 3-tetrafluoro-1-propanol (TFP), 2, 2, 2-trifluoroethanol (TFE), 2, 2, 3, 3, 4, 4, 5, 5-octafluoro-1-pentanol (OFP) mixed with H2O in the ratio of 1:1 did not afford higher yields (entries 6–8). Reactions carried out at 25 ℃, 45 ℃ and 80 ℃, respectively, gave 69%–72% yield of 5d (entries 9–11). A loading of 1.5 equiv. of IBX was not enough for the transformation, as seen in the significantly lower yield (entry 12).
With the optimized conditions in hand (Table 1, entry 1), we first explored the scope of this IBX promoted intermolecular oxidative spiro-cyclization reaction with representative chiral α-amino amides bearing different arene sulfonyl substitutions at the α-position. As shown in Scheme 4, α-AA derivatives bearing EtO2C- (1a), CF3- (1b) and Cl- (1c) group at the 4-position of the aryl group, as well as p-toluene sulfonyl (Ts-) substitution (1d), gave target products 5a–5d in 74%-91% yields with ca. 7:1 dr. However, 4-OMe- and 4-nitrobenesulfonyl (Nos-) substituted starting materials 1e and 1f only gave 5e and 5f in 38% and 61% yields, respectively, with ca. 8:1 to 5:1 dr. Considering the price of various of arylsulfonyl substitutions, we used the Ts- group to investigate the influence of other substituents at the α-position on the conversion. N-Ts substituted amide derivatives (1g–1i) derived from Phe, Leu and Val proceeded in good yields (5g–5i, 65%–80%) and acceptable diastereoselectivities (dr: 2:1-10:1) under optimized conditions. To our delight, the transformation also tolerated 8-isoquinolinyl (1j), 5-quinolinyl (1j) and 1-naphthalenyl (1l) groups, which afforded the corresponding five-membered spiro compounds 5j–5l. These examples further expanded the scope of this heterocyclic spiro-annulations to construct more structurally diverse N- and O-containing spirocompounds. Notably, compounds 5j and 5k are representative examples of 5, 6-dearomatization of quinolines complementary to the 7, 8-dearomatized ones, which are far from being explored. Preassemble of the hydroxyl on naphthaline to facilitate later spiroannulation [63], as practised by Chang et al., was not required in our process. This further highlights the advantages of this in situ oxidative cyclization strategy.
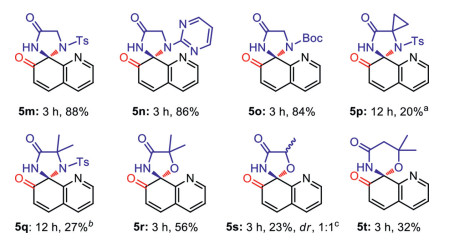
To further investigate the scope, some α-AA derivatives derived from nonchiral amino acids were employed to the reaction (Scheme 5). Starting materials 1m–1o derived from Gly with three kinds of amino protecting groups including Ts-, 2-pyrimidinyl, and Boc- could generate desired products 5m–5o in 84%-88% yield after 3 h. However, it seems that the increasing the steric hindrance at the α-position by gem-dimethyl could hamper the smooth nucleophilic addition, as compounds 1p and 1q only afforded target products 5p and 5q in 20% and 27% yields, respectively. Pleasingly, α-dimethyl-α-hydroxyl amide 1r gave 5r in a moderate yield of 56%. In contrast, α-methyl-α-hydroxyl amide 1s showed a lower reactivity and gave product 5s in the yield of 23%, along with 11% oxidation product 2-oxo-N-(quinolin-8-yl)propanamide [64]. Preliminary exploration on the six-membered spiro product starting from 1t only gave 5t in 32% yield.
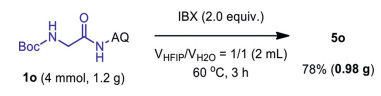
The HFIP solvent, which could be recovered under reduced pressure after the reaction, contributes positively to the E-factor of these conversion [65], which would be prerequisites of these reactions performed on large scale. The operational simplicity of this 7, 8-dearomatization reaction allowed rapid access gem-diamine-containing spiro compound 5o, which has not been accessed by conventional strategies, from readily available 1o after 3 h (Scheme 6). In addition, the Boc-protecting group could be facile removed to reveal the amine for subsequent functionalization.
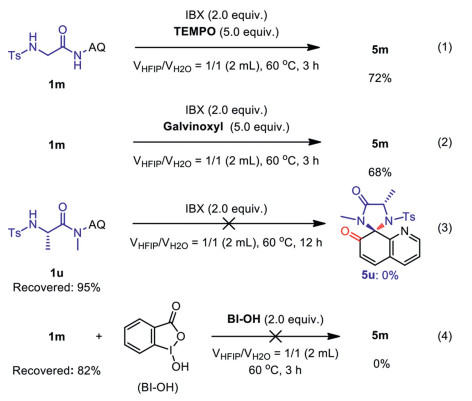
Many works have been presented a radical or nucleophilic mechanistic pathway for the IBX-mediated ring closures of anilides and related systems to N-heterocycles [55, 66–68]. Based on these pioneering works, we suspected if our transformation may also undergo a radical pathway. The reactions in the presence of 5.0 equiv. of TMPO and galvinoxyl radical traps were carried out under the standard conditions, respectively. As a result, the reactions proceeded smoothly without being much suppressed (Scheme 7, Eqs. 1 and 2). We conclude accordingly that SET process may be not involved. In addition, no reaction was observed with N-methyl amide starting material 1u (Scheme 7, Eq. 3). This control experiment indicated that the NH amide moiety plays a key role for the reactivity. In addition, when hydroxy benziodoxolone (BI-OH, I(III)) was employed in the cyclization of 1m, no reaction was observed (Scheme 7, Eq. 4).
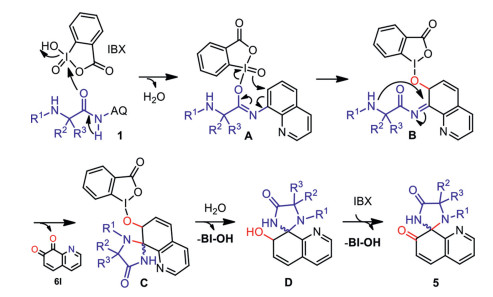
A plausible mechanism is outlined in Scheme 8. This IBX-promoted spirocyclization reaction probably starts with nucleophilic attack of amide of 1 to the iodo center of IBX to form iodoimidate intermediate A [55, 69]. Then, intramolecular nucleophilic attack of the oxo group on the iodine center onto the ortho-carbon of AQ group triggers 7, 8-dearomatization of aniline and O‒I bond cleavage to form B [69–71]. Subsequently, intramolecular nucleophilic attack of the side chain amino group on the imine carbon leads to cyclization to afford five-membered spirocycle C [72–74]. Followed by hydrolysis and oxidation by an extra molecule of IBX, the desired spiroquinlinones 5 is generated by releasing of BI-OH [75]. In addition, the hydrolysis reaction leads to the AQ group decomposes from the imine intermediate B, producing the byproduct quinoline-7, 8-dione 6l with the help of IBX [75].
As mentioned above, the two-fold oxidation processes accounts for the requirement of 2 equiv. of IBX oxidant. Notably, commercially available trivalent organoiodine reagent BI-OH showed no reactivity in a separated control experiment (Eq. 4). Furthermore, a molecular ion peak of m/z 264.9 was detected by MS spectroscopy in the reaction mixture under the standard conditions, which corresponds to the molecular weight of the BI-OH, while the molecular ion peak of 2-iodobenzoic acid (2-IBA) was not detected, further indicating that the hypervalent iodine reagent IBX is reduced to trivalent iodine IIII instead of monovalent iodine II in the process.
In summary, an IBX-mediated intramolecular oxidative spirocyclization of α-AA analogues bearing NHTs side-chains was developed for rapid access to spirocyclic quinolinones in moderate to good yields under mild conditions. This tandem reaction features an unusual 7, 8-dearomatization of quinolines, delivering unique spiro[imidazolidine-2, 8′-quinoline]-4, 7′-dione 5 [76]. Among them, two kinds of novel six-membered aza- and oxa-heterocycle-containing spiro quinoline compounds are synthesized for the first time. The synthesized spirocompounds may open the door to a series of spiroquinolinones of potential interest in synthetic and medicinal chemistry. Mechanistic studies suggest that the reaction proceeds by intramolecular nucleophilic cyclization of the oxidatively generated o-iminoquinone intermediate by the pendant amino group at the side chain. Further studies on the asymmetric synthesis are currently underway in our laboratory.
We declare that we have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the National Natural Science Foundation of China (Nos. 22101074, 21877206, and 21772032), the 111 Project (No. D17007), Excellent Youth Foundation of Henan Scientific Committee (No. 222300420012), China Postdoctoral Science Foundation (No. 2019M660173), the Natural Science Foundation of Henan Province (No. 202300410233).
Supplementary material associated with this article can be found, in the online version, at doi:
C. Zheng, S.L. You, ACS Central Sci. 7 (2021) 432–444. doi: 10.1021/acscentsci.0c01651
K. Sun, G. Li, S. Guo, Z. Zhang, G. Zhang, Org. Biomol. Chem. 19 (2021) 375–378. doi: 10.1039/d0ob02210a
F.L. Zeng, X.L. Chen, K. Sun, et al., Org. Chem. Front. 8 (2021) 760–766. doi: 10.1039/d0qo01410a
Z.J. Zuo, J. Wang, J.J. Liu, Y.Y. Wang, X.J. Luan, Angew. Chem. Int. Ed. 59 (2020) 653–657. doi: 10.1002/anie.201909557
R.C. McAtee, E.A. Noten, C.R.J. Stephenson, Nat. Commun. 11 (2020) 2528–2536. doi: 10.1038/s41467-020-16369-4
K. Sun, G. Li, Y. Li, et al., Adv. Synth. Catal. 362 (2020) 1947–1954. doi: 10.1002/adsc.202000040
Y. Chen, Y.J. Chen, Z. Guan, Y.H. He, Tetrahedron 75 (2019) 130763. doi: 10.1016/j.tet.2019.130763
Y. Liu, Q.L. Wang, B.Q. Xiong, et al., Synlett 29 (2018) 2396–2403. doi: 10.1055/s-0037-1609948
W. Hu, H. Wang, L. Bai, J. Liu, X. Luan, Org. Lett. 20 (2018) 880–883. doi: 10.1021/acs.orglett.8b00014
L.J. Wu, F.L. Tan, M. Li, R.J. Song, J.H. Li, Org. Chem. Front. 4 (2017) 350–353. doi: 10.1039/C6QO00691D
R. Song, Y. Xie, Chin. J. Chem. 35 (2017) 280–288. doi: 10.1002/cjoc.201600846
X. Ma, J.J. Farndon, T.A. Young, N. Fey, J.F. Bower, Angew. Chem. Int. Ed. 56 (2017) 14531–14535. doi: 10.1002/anie.201708176
M. Li, R.J. Song, J.H. Li, Chin. J. Chem. 35 (2017) 299–302. doi: 10.1002/cjoc.201600749
N. Hegmann, L. Prusko, M.R. Heinrich, Org. Lett. 19 (2017) 2222–2225. doi: 10.1021/acs.orglett.7b00676
D.M. Kuznetsov, A.G. Kutateladze, J. Am. Chem. Soc. 139 (2017) 16584–16590. doi: 10.1021/jacs.7b07598
X. Zhang, Y. Cong, G. Lin, et al., Chin. J. Org. Chem. 36 (2016) 2513–2529. doi: 10.6023/cjoc201605034
W. Wei, H. Cui, D. Yang, et al., Green Chem. 19 (2017) 5608–5613. doi: 10.1039/C7GC02330H
D. Li, X.C. Chen, W. Gao, Synth. Stuttg. 52 (2020) 3337–3355. doi: 10.1055/s-0040-1707206
X. Yan, L. Ge, M. Castiñeira Reis, S.R. Harutyunyan, J. Am. Chem. Soc. 142 (2020) 20247–20256. doi: 10.1021/jacs.0c09974
D. Wang, Z. Wang, Z. Liu, et al., Org. Lett. 21 (2019) 4459–4463. doi: 10.1021/acs.orglett.9b01247
D. Wang, Y. Jiang, L. Dong, et al., J. Org. Chem. 85 (2020) 5027–5037. doi: 10.1021/acs.joc.0c00314
M. Zurro, S. Asmus, S. Beckendorf, C. Mück-Lichtenfeld, O.G. Mancheño, J. Am. Chem. Soc. 136 (2014) 13999–14002. doi: 10.1021/ja507940k
J. Jeong, J. Heo, D. Kim, S. Chang, ACS Catal. 10 (2020) 5023–5029. doi: 10.1021/acscatal.0c00884
S.R. Tamang, A. Singh, D.K. Unruh, M. Findlater, ACS Catal. 8 (2018) 6186–6191. doi: 10.1021/acscatal.8b01166
F. Zhang, H. Song, X. Zhuang, C.H. Tung, W. Wang, J. Am. Chem. Soc. 139 (2017) 17775–17778. doi: 10.1021/jacs.7b11416
V.D. Cao, S.H. Mun, S.H. Kim, et al., Org. Lett. 22 (2020) 515–519. doi: 10.1021/acs.orglett.9b04275
N. Gandhamsetty, S. Joung, S.W. Park, S. Park, S. Chang, J. Am. Chem. Soc. 136 (2014) 16780–16783. doi: 10.1021/ja510674u
H.J. Zhang, Z.P. Yang, Q. Gu, S.L. You, Org. Lett. 21 (2019) 3314–3318. doi: 10.1021/acs.orglett.9b01060
Z.P. Yang, Q.F. Wu, W. Shao, S.L. You, J. Am. Chem. Soc. 137 (2015) 15899–15906. doi: 10.1021/jacs.5b10440
Q.F. Xu Xu, X. Zhang, S.L. You, Org. Lett. 21 (2019) 5357–5362. doi: 10.1021/acs.orglett.9b02034
M. Shetty, H. Huang, J.Y. Kang, Org. Lett. 20 (2018) 700–703. doi: 10.1021/acs.orglett.7b03829
S.G. Wang, W. Zhang, S.L. You, Org. Lett. 15 (2013) 1488–1491. doi: 10.1021/ol4002416
X. Song, R.J. Yan, W. Du, Y.C. Chen, Org. Lett. 22 (2020) 7617–7621. doi: 10.1021/acs.orglett.0c02828
N. De, D. Ko, S.Y. Baek, et al., ACS Catal. 10 (2020) 10905–10913. doi: 10.1021/acscatal.0c03014
R. Kumar, S. Chaudhary, R. Kumar, et al., J. Org. Chem. 83 (2018) 11552–11570. doi: 10.1021/acs.joc.8b01520
Z. Kang, D. Zhang, W. Hu, Org. Lett. 19 (2017) 3783–3786. doi: 10.1021/acs.orglett.7b01664
L.Y. Xie, Y. Duan, L.H. Lu, et al., ACS Sustain. Chem. Eng. 5 (2017) 10407–10412. doi: 10.1021/acssuschemeng.7b02442
B. Zhang, L. Huang, S. Yin, et al., Org. Lett. 19 (2017) 4327–4330. doi: 10.1021/acs.orglett.7b01996
Y.Y. Zhou, J. Li, L. Ling, et al., Angew. Chem. Int. Ed. 52 (2013) 1452–1456. doi: 10.1002/anie.201207576
S. Sowmiah, J.M.S.S. Esperança, L.P.N. Rebelo, C.A.M. Afonso, Org. Chem. Front. 5 (2018) 453–493. doi: 10.1039/c7qo00836h
Z.P. Yang, R. Jiang, C. Zheng, S.L. You, J. Am. Chem. Soc. 140 (2018) 3114–3119. doi: 10.1021/jacs.8b00136
D. Sarkar, N. Rout, Org. Lett. 21 (2019) 4132–4136. doi: 10.1021/acs.orglett.9b01322
H.J. Lee, X. Huang, S. Sakaki, K. Maruoka, Green Chem. 23 (2021) 848–855. doi: 10.1039/d0gc03912h
F. Ballaschk, S.F. Kirsch, Green Chem. 21 (2019) 5896–5903. doi: 10.1039/c9gc02605c
Y.N. Ma, C.Y. Guo, Q. Zhao, J. Zhang, X. Chen, Green Chem. 20 (2018) 2953–2958. doi: 10.1039/c8gc01057a
S.V. Kohlhepp, T. Gulder, Chem. Soc. Rev. 45 (2016) 6270–6288. doi: 10.1039/C6CS00361C
Y. Duan, S. Jiang, Y. Han, B. Sun, C. Zhang, Chin. J. Org. Chem. 36 (2016) 1973–1984. doi: 10.6023/cjoc201605007
K. Ouyang, W. Hao, W.X. Zhang, Z. Xi, Chem. Rev. 115 (2015) 12045–12090. doi: 10.1021/acs.chemrev.5b00386
J. Chen, H. Qu, J. Peng, C. Chen, Chin. J. Org. Chem. 35 (2015) 937–946. doi: 10.6023/cjoc201501004
V.V. Zhdankin, P.J. Stang, Chem. Rev. 108 (2008) 5299–5358. doi: 10.1021/cr800332c
A.N. French, S. Bissmire, T. Wirth, Chem. Soc. Rev. 33 (2004) 354–362. doi: 10.1039/b310389g
Z. Zhang, X. Song, G. Li, et al., Chin. Chem. Lett. 32 (2021) 1423–1426. doi: 10.1016/j.cclet.2020.11.001
M. Song, Z. Zhang, D. Zheng, et al., Chin. J. Org. Chem. 40 (2020) 2433–2441. doi: 10.6023/cjoc202001007
W. Gao, Y. Wan, Z. Zhang, et al., Green Chem. 22 (2020) 7955–7961. doi: 10.1039/d0gc02777d
Z. Zhang, X. Li, M. Song, et al., J. Org. Chem. 84 (2019) 12792–12799. doi: 10.1021/acs.joc.9b01362
Z. Zhang, D. Zheng, Y. Wan, et al., J. Org. Chem. 83 (2018) 1369–1376. doi: 10.1021/acs.joc.7b02880
J. Dong, M. Jia, X. Xu, Chin. Chem. Lett. 32 (2021) 1831–1833. doi: 10.1016/j.cclet.2021.01.034
Z. Liu, H. Ji, W. Gao, et al., Chem. Commun. 53 (2017) 6259–6262. doi: 10.1039/C7CC02391J
S.Y. Zhang, Q. Li, G. He, W.A. Nack, G. Chen, J. Am. Chem. Soc. 137 (2015) 531–539. doi: 10.1021/ja511557h
M. Tobisu, K. Nakamura, N. Chatani, J. Am. Chem. Soc. 136 (2014) 5587–5590. doi: 10.1021/ja501649a
D.R. Kronenthal, C.Y. Han, M.K. Taylor, J. Org. Chem. 47 (1982) 2765–2768. doi: 10.1021/jo00135a016
Z.G. Zhang, X.Y. Cao, G. Wang, G.S. Zhang, X.J. Zhang, Green Chem. 24 (2022) 3035–3041. doi: 10.1039/d2gc00395c
E. Lee, Y. Hwang, Y.B. Kim, D. Kim, S. Chang, J. Am. Chem. Soc. 143 (2021) 6363–6369. doi: 10.1021/jacs.1c02550
F.X. Felpin, Tetrahedron Lett. 48 (2007) 409–412. doi: 10.1016/j.tetlet.2006.11.073
R.A. Sheldon, Green Chem. 9 (2007) 1273–1283. doi: 10.1039/b713736m
V.V. Zhdankin, J. Org. Chem. 76 (2011) 1185–1197. doi: 10.1021/jo1024738
U. Ladziata, V.V. Zhdankin, Arkivoc (2006) 26–58. doi: 10.3998/ark.5550190.0007.903
K.C. Nicolaou, P.S. Baran, R. Kranich, et al., Angew. Chem. Int. Ed. 40 (2001) 202–206. doi: 10.1002/1521-3773(20010105)40:1<202::AID-ANIE202>3.0.CO;2-3
K.C. Nicolaou, K. Sugita, P.S. Baran, Y.L. Zhong, J. Am. Chem. Soc. 124 (2002) 2221–2232. doi: 10.1021/ja012125p
K.C. Nicolaou, P.S. Baran, Y.L. Zhong, K. Sugita, J. Am. Chem. Soc. 124 (2002) 2212–2220. doi: 10.1021/ja012124x
N. Thrimurtulu, A. Dey, K. Pal, et al., ChemistrySelect 2 (2017) 7251–7254. doi: 10.1002/slct.201701388
Z. Hu, M. Zhang, Q. Zhou, X. Xu, B. Tang, Org. Chem. Front. 7 (2020) 507–512. doi: 10.1039/c9qo01333d
G. Ramachandran, K.I. Sathiyanarayanan, Tetrahedron Lett. 54 (2013) 6758–6763. doi: 10.1016/j.tetlet.2013.10.009
C.R. Reddy, S.K. Prajapti, K. Warudikar, R. Ranjan, B.B. Rao, Org. Biomol. Chem. 15 (2017) 3130–3151. doi: 10.1039/C7OB00405B
A. Duschek, S.F. Kirsch, Angew. Chem. Int. Ed. 50 (2011) 1524–1552. doi: 10.1002/anie.201000873
U. Gruseck, M. Heuschmann, Tetrahedron Lett. 28 (1987) 6027–6030. doi: 10.1016/S0040-4039(00)96855-2

Scheme 4 Extension scope of chiral α-amino amide analogs. Isolated yield on 0.2 mmol scale under the standard conditions. The dr value was determined by 1H NMR, and the sample is taken from a portion of the mixture of all products. a 21% of 1i was recovered. b Reaction was performed at 100 ℃. c 9% of quinoline-7, 8-dione (6l) was obtained simultaneously.

Scheme 5 Extension scope of other α-amino amide analogs. Isolated yield on 0.2 mmol scale under the standard conditions. The dr value was determined by 1H NMR, and the sample is taken from a portion of the mixture of all products. a 35% of 1p was recovered. b 46% of 1q was recovered.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:




