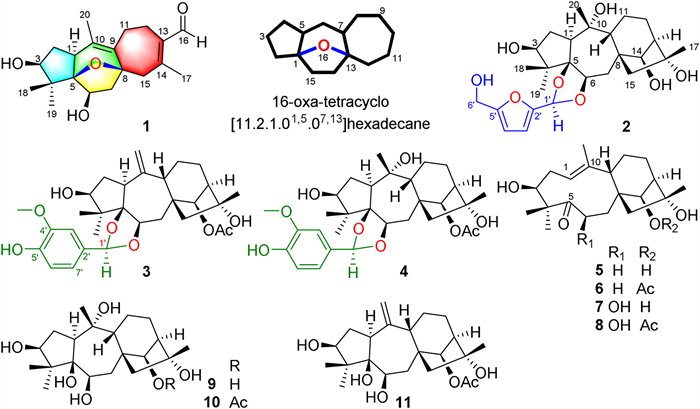
Figure 1.
Chemical structures of compounds 1−11 and the nomenclature of the unprecedented tetracyclic ring system in 1.
Rhodauricanol A, an analgesic diterpenoid with an unprecedented 5/6/5/7 tetracyclic system featuring a unique 16-oxa-tetracyclo[11.2.1.01,5.07,13]hexadecane core from Rhododendron dauricum
Yuanyuan Feng , Suqin Zha , Hanqi Zhang , Biao Gao , Guijuan Zheng , Pengfei Jin , Yingyi Chen , Guangmin Yao

Polycyclic diterpenoids such as taxol with a 6/8/6 tricyclic carbon skeleton are a potential resource for drug discovery [1]. Ericaceae plants are famous for their structurally diverse diterpenoids, heretofore, approximately 470 diterpenoids belonging to 28 carbon skeletons have been reported [2-4]. Due to their highly oxygenated and polycyclic architectures, the total synthesis of Ericaceae diterpenoids is an enduring research hotspot. To date, the total synthesis of six grayanane [5-10], a rhodomollane [11], and two mollebenzylane [12,13] diterpenoids have been accomplished, and the synthetic studies towards kalmanol [14] and pierisformoside C [15] were also described.
Severe pains, especially cancer pains, seriously affect people's quality of life. As the gold standard for comparison of other opioids, morphine is used for acute and chronic pain and plays an irreplaceable role in patients with moderate and severe cancer pain. However, the main side effects of morphine are drug resistance and addiction [16]. Thus, it is urgent to search for potent analgesics without addiction and tolerance.
In a continuing search for structurally intriguing analgesics from the medicinal plants [17], a novel diterpenoid with an unprecedented carbon skeleton, rhodauricanol A (1), five new grayanane-derived diterpenoids, dauricanols A−E (2−6), and five known ones (7−11) (Fig. 1) were isolated from the flowers of Rhododendron dauricum L. (Ericaceae), which have been used for sedation and hypnosis. Rhodauricanol A (1) possesses a unique 5/6/5/7 tetracyclic ring system featuring a 16-oxa-tetracyclo[11.2.1.01,5.07,13]hexadecane core. Dauricanols A−C (2−4) are the first 1,3-dioxolane conjugates of grayanane diterpenoids and 5-hydroxymethylfurfural and vanillin, respectively, and dauricanols D (5) and E (6) represent the first examples of 6-deoxy-1,5-seco-grayanane diterpenoid. All the isolates showed potent analgesic effects, and 3 and 4 showed more potent analgesic activities than morphine at a dose of 0.04 mg/kg. Herein, the structural elucidation, plausible biosynthetic pathways, and analgesic activities were described.
Rhodauricanol A (1), colorless needles, had a molecular formula of C20H28O4 as deduced by the HRESIMS ([M + Na]+ m/z 355.1885, calcd. for 355.1885) and 13C NMR data, indicating seven degrees of unsaturation. The 1H NMR data (Table S1 in Supporting information) of 1 showed resonances attributed to an aldehyde proton at δH 10.05 (s, H-16), two oxygenated methines at δH 3.47 (d, J = 6.0 Hz, H-3) and 4.28 (dd, J = 7.6, 3.4 Hz, H-6), and four methyl groups at δH 1.12 (s, H3-19), 1.28 (s, H3-18), 1.58 (s, H3-20), and 2.29 (s, H3-17). The 13C NMR and DEPT spectra (Table S2 in Supporting information) revealed 20 carbon signals, including four methyls, five methylenes, three methines (two oxygenated), a quaternary carbon, two oxygenated tertiary carbons, two tetrasubstituted endocyclic double bonds, and an aldehyde. Two double bonds and an aldehyde account for three degrees of unsaturation, and the remaining four degrees of unsaturation suggested a tetracyclic ring system in 1.
1H−1H COSY revealed three spin-coupling systems (Fig. 2): (a) CH-1−CH2-2−CH-3, (b) CH-6−CH2-7, and (c) CH2-11−CH2-12. The HMBC correlations of two gem-dimethyl (H3-18/H3-19) to C-3/C-4/C-5 indicated the direct connections of C-3/C-5/C-18/C-19 to the quaternary carbon C-4. The connection of C-1 and C-6 through the oxygenated tertiary carbon C-5 was determined by the HMBC correlations from H-6 to C-1 and H2-7 to C-5, forming the A-ring of 2,2-dimethyl-cyclopentan-1-ol in 1. The HMBC correlations from H3-20 to C-1/C-9/C-10 established the connection of C-1 and C-9 via C-10. The connection of C-9 to C-7 through an oxygenated tertiary carbon C-8 was determined by the HMBC correlations from H2-7 to C-8/C-9, constructing the B-ring of 4-methyl-4-cycloheptene-1-ol. The C-ring of 2-methyl-1-cycloheptene-1-carbaldehyde in 1 was architected by the HMBC correlations from H-11 to C-8, from H-12 to C-9/C-13/C-14, from H-16 to C-12/C-13, from H2-15 to C-8/C-9, and from H3-17 to C-13/C-14/C-15. The large chemical shift of C-5 (δC 95.5) and C-8 (δC 84.1) and the rest of one degrees of unsaturation suggested the presence of an oxygen bridge between C-5 and C-8. Thus, the planar structure of 1 bearing an unprecedented 16-oxa-tetracyclo [11.2.1.01,5.07,13]hexadecane motif was established (Fig. 2).

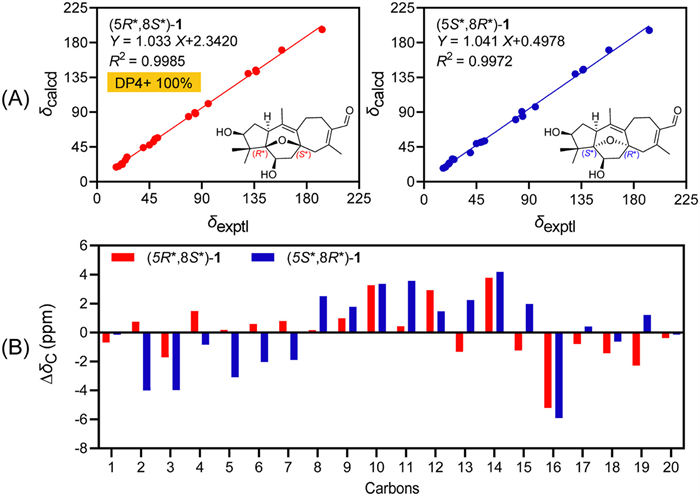
The relative configuration of 1 was established by NOESY data analysis and 13C NMR-DP4+ analysis [18,19]. H-1 was randomly assigned as α-orientation. The NOESY correlations of H-1/H-6, H-1/H3-19, and H3-19/H-3 defined the α orientations of H-3, H-6, and H3-19. The geometry of Δ9(10) and Δ13(14) double bonds were assigned as Z and E, respectively, by the NOESY correlations of H-11β/H3-20 and H-16/H3-17. To determine the orientation of 5,8-epoxy moiety, the theoretical 13C NMR calculations and DP4+ probability analysis of two potential isomers, (5R*,8S*)-1 and (5S*,8R*)-1, were performed using the gauge independent atomic orbital (GIAO) method at the mPW1PW91/6-311G(d,p) level with the Gaussian 09 software [20-22]. The smaller relative errors between experimental and the calculated 13C NMR data (Fig. 3) and the DP4+ probability analysis revealed the 5R*,8S*-configuration of 1 with a high probability of 100% (Table S3 in Supporting information) [18].
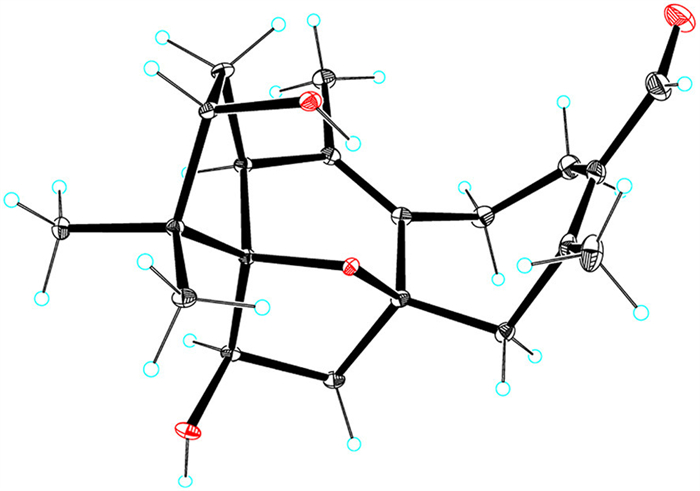
To determine the absolute configuration of 1, the TDDFT-based ECD calculation for (1S,3S,5R,6R,8S)-1 and its enantiomer was performed [23,24]. A good qualitative agreement between the experimental and calculated ECD curves (Fig. S1 in Supporting information) of 1 determined its absolute configuration as 1S,3S,5R,6R,8S. Finally, single-crystal X-ray diffraction analysis using Cu Kα radiation with a Flack parameter [25] of 0.05(4) further validated the structure and absolute configuration of 1 (Fig. 4).
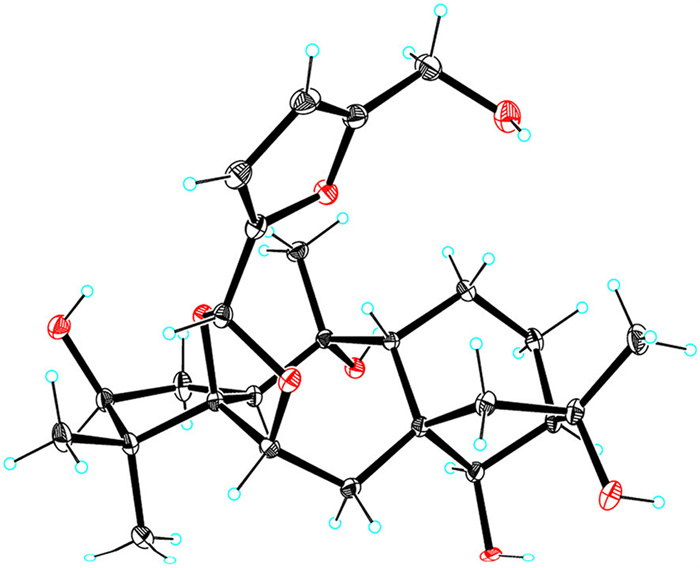
A molecular formula of C26H38O8 was assigned to dauricanol A (2) based on the HRESIMS at m/z 501.2465 [M + Na]+ (calcd. for C26H38O8Na, 501.2464) and 979.5040 [2M + Na]+ (calcd. for C52H76O16Na, 979.5031) and 13C NMR data. The NMR data (Tables S1 and S2) of 2 showed some similarities to those of grayanotoxin Ⅲ (9) [26], co-isolated grayanane diterpenoid in this study. The major differences were the presence of an acetal group (δH 5.70, s, H-1′), two trisubstituted double bonds (δH 6.45, d, H-3′; 6.30, d, H-4′; δC 111.9, C-3′; 109.1, C-4′; 150.5, C-2′; 157.0, C-5′), and an oxymethylene (δH 4.49, s, H2-6′) in 2, which was determined to be a (5-(hydroxymethyl)-2-furanyl)methane-1,1-diyl moiety by the detailed 2D NMR analysis (Fig. S2 in Supporting information). The deshielding of C-5 (δC 93.6) and C-6 (δC 78.3) in 2 compared to 9 (δC 84.6, C-5; 74.2, C-6) and the HMBC correlations from H-1′ to C-5 and C-6 suggested the connection of the additional (5-(hydroxymethyl)-2-furanyl)methane-1,1-diyl moiety to C-5 and C-6 via two ether bonds, forming a distinctive 1,3-dioxolane ring. H-1 was randomly assigned as α-orientation. The NOESY correlations (Fig. S2) of H-1/H3-19, H3-19/H-6, and H-6/H-1′ established α-orientation of H-1′. The absolute configuration of 2 was defined as 1S,3S,5R,6R,8S,9R,10R,13R,14R,16R,1′R by ECD calculation (Fig. S3 in Supporting information) and single crystal X-ray diffraction analysis with a Flack parameter [25] of −0.03(2) (Fig. 5).
The molecular formula of dauricanol B (3) was determined to be C30H40O8 by the HRESIMS ion at m/z [M + Na]+ 551.2616 (calcd. for C30H40O8Na, 551.2621) and [2M + Na]+ 1079.5343 (calcd. for C60H80O16 Na, 1079.5344) and 13C NMR data. The NMR data of 3 (Tables S1 and S2) resembled those of rhodomicranol A, which was firstly isolated from Rhododendron micranthum [27], and the major difference was an additional O-CH3 (δH 3.83, s; δC 56.5) in 3. Thus, 3 is an O-methylated derivative of rhodomicranol A. HMBC correlation from OCH3 to C-4′ and the NOESY correlation of H-3′ and OCH3 suggested the location of OCH3 at C-4′ (Fig. S4 in Supporting information). H-1 was randomly assigned as α-orientation. NOESY correlations of H-1/H3-19, H3-19/H-6, and H-6/H-1′ revealed the α-orientation of H-1′. The absolute configuration of 3 was determined to be 1S,3S,5R,6R,8S,9S,13R,14R,16R,1′R by the ECD calculation (Fig. S5 in Supporting information).
Dauricanol C (4) was assigned a molecular formula of C30H42O9 by the 13C NMR data and HRESIMS ions at m/z [M + Na]+ 569.2717 (calcd. for C30H42O9Na, 569.2727) and [2M + Na]+ 1115.5578 (calcd. for C60H84O18Na, 1115.5555). The NMR data (Tables S2 and S4 in Supporting information) of 4 were similar to those of 3, except for a methyl group (δH 1.72, s; δC 26.9, CH3-20) and an oxygenated tertiary carbon (δC 78.3, C-10) in 4, replacing an exocyclic double bond (δH 5.05, 5.07, s, H2-20; δC 152.0, C-10; 112.4, C-20) in 3. Thus, 4 is a hydrated derivative of 3. The HMBC correlations (Fig. S6 in Supporting information) from H3-20 to C-1/C-9/C-10 supported the above deduction. The NOESY correlation (Fig. S6) of H-2β and H3-20 suggested H3-20 was β-oriented. ECD calculation (Fig. S7 in Supporting information) defined the 1S,3S,5R,6R,8S,9R,10R,13R,14R,16R,1′R-configuration of 4.
The molecular formula of dauricanol D (5) was determined as C20H32O4 by the 13C NMR data and HRESIMS ions at m/z [M + Na]+ 359.2199 (calcd. for C20H32O4Na, 359.2198) and [2M + Na]+ 695.4574 (calcd. for C40H64O8Na, 695.4499). The NMR data (Tables S2 and S4) of 5 was close to the co-isolated 1,5-seco-grayanotoxin (7) [28]. The major difference was a methylene (δH 2.56, 3.01, H2-6; δC 36.3, C-6) in 5, replacing an oxygenated methine (δH 4.88, br. s, H-6; δC 70.5, C-6) in 7. Thus, 5 should be a 6-deoxy derivative of 7, which was supported by the 1H–1H COSY correlations (Fig. S8 in Supporting information) of CH2-6 and CH2-7. The structure and 3S,8S,9S,13R,14R,16R-configuration of 5 was verified by single-crystal X-ray diffraction analysis (Fig. 6) with a Flack parameter [25] of 0.01(4).
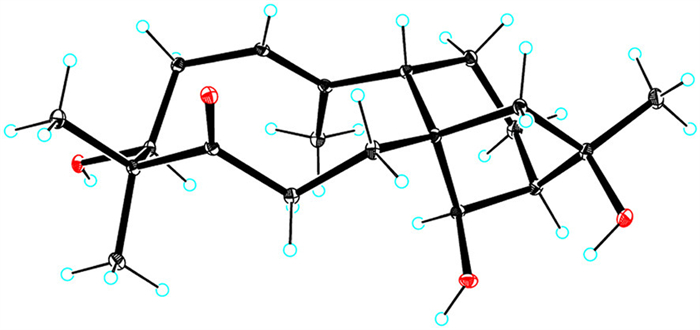
Dauricanol E (6) had a molecular formula of C22H34O5 as disclosed by the HRESIMS ions at m/z [M + Na]+ 401.2324 (calcd. for C22H34O5Na, 401.2304) and [2M + Na]+ 779.4724 (calcd. for C44H68O10Na, 779.4710) and 13C NMR data. The NMR data (Tables S2 and S4) of 6 and 5 were closely comparable, except for an O-acetyl group (δH 2.07, s; δC 172.5, 21.5) and the deshielding of C-14 (δC 82.6) in 6 compared to 5 (δC 79.4). Hence, 6 was a 14-O-acetylated derivative of 5, which was proved by the 1H–1H COSY correlation of H-13 and H-14 and HMBC correlation of H-14 to the acetyl (Fig. S9 in Supporting information). Single-crystal X-ray diffraction analysis (Fig. 7) with a Flack parameter [25] of 0.04(7) ultimately defined the 3S,8S,9S,13R,14R,16R-absolute configuration of 6.
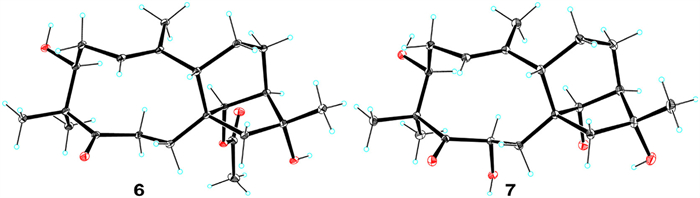
1,5-seco-Grayanane diterpenoids are rare in nature, heretofore, only eight 1,5-seco-grayanane diterpenoids have been reported [4,17]. Dauricanols D (5) and E (6) represent the first examples of 6-deoxy-1,5-seco-grayanane diterpenoid. This is also the first time to report 1,5-seco-grayanane diterpenoids from this plant.
Five known compounds were identified to be 1,5-seco-grayanotoxin (7) [28] and grayanotoxins XXI (8) [29], Ⅲ (9) [26], Ⅰ (10) [30], and Ⅳ (11) [30], respectively, by spectroscopic data analysis and comparison with the reported data. The structure and absolute configuration of 1,5-seco-grayanotoxin (7) was finally assigned by single-crystal X-ray diffraction analysis (Fig. 7) with a Flack parameter [25] of −0.07(5) for the first time.
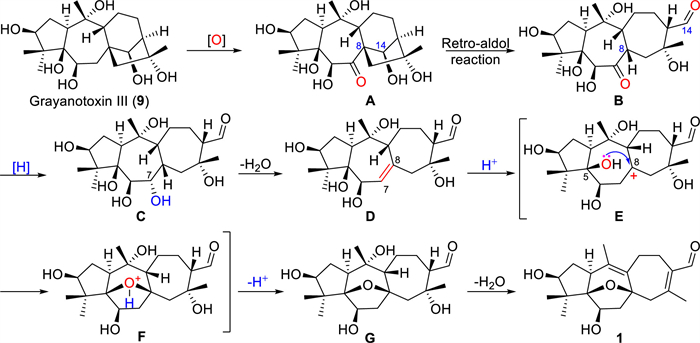
Rhodauricanol A (1) possesses an unprecedented 5/7/7 tricyclic diterpene carbon skeleton, and the name “rhodauricane” was suggested for this new diterpene skeleton, enriching the diterpene skeleton diversity of natural products. The plausible biosynthetic pathway of 1 (Scheme 1) could be tracked back to the co-isolated grayanotoxin Ⅲ (9) [26]. Under the condition of an enzyme-mediated oxidization, 9 is oxidized to a 7-ketone intermediate A. The carbon bond of C-8 and C-14 in A is cleaved by the retro-aldo reaction to produce the key tricyclic intermediate B, which is reduced and then dehydrated to form the ∆7(8) intermediate D. Under the catalysis of acid enzyme, 5-OH attacks the C-8 carbocation in E to yield the 5,8-epoxy intermediate F. Finally, 1 is generated by the dehydration of G.
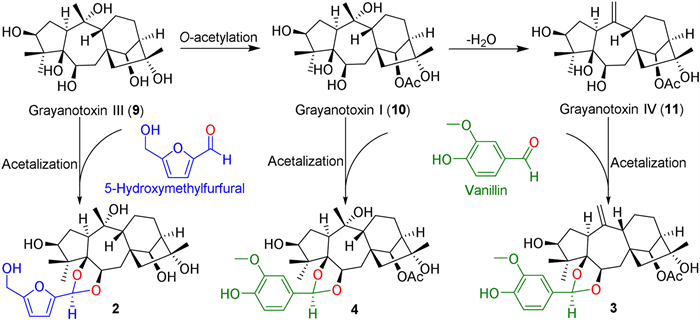
Dauricanols A−C (2−4) are the first 1,3-dioxolane conjugates of grayanane diterpenoids and 5-hydroxymethylfurfural and vanillin, respectively. Dauricanol A (2) is produced by an acetalation reaction (Scheme 2) of the 5,6-dihydroxyl unit in 9 with 5-hydroxymethylfurfural which is derived from the dehydration of hexose [31] and widely exists in plants [32]. Similarly, dauricanols B (3) and C (4) are derived from the condensation of vanillin [33] and grayanotoxins Ⅰ (10) and Ⅳ (11), respectively (Scheme 2).
Since the flowers of R. dauricum are used for sedation and hypnosis, and three known grayanane diterpenoids 9−11 had been reported to possess analgesic activities, the other isolates 1−8 were evaluated for their analgesic activities by the HOAc-induced writhing test in mice [4]. This animal experiment was approved by the Laboratory Animal Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (approval number 2018-S748). Results revealed that 1−8 showed significant analgesic effects (Fig. S10 in Supporting information). Rhodauricanol A (1) with a new diterpene carbon skeleton showed significant analgesic effects at lower doses of 1.0 and 0.2 mg/kg with inhibition rates of 65.2% and 44.2%, respectively. Interestingly, 3 and 4, 1,3-dioxolane conjugates of grayanane diterpenoids and vanillin, exhibited more potent activity than morphine even at a lower dose of 0.04 mg/kg with inhibition rates of 62.8% and 53.2%, respectively. 1,5-seco-Grayanane diterpenoid 5 bearing a 14β-OH showed more potent analgesic effect than 6 with a 14β-OAc at doses of 5.0 and 1.0 mg/kg. While, 5 and 6 without a 6β-OH displayed more potent analgesic activity than 7 and 8 with a 6β-OH at a dose of 5.0 mg/kg. Thus, 14β-OAc and 6β-OH groups may be the deactivating groups of 1,5-seco grayanane diterpenoids.
These findings enriched the diterpene carbon skeleton diversity and provided clues to design potent analgesics.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Fund of State Key Laboratory of Phytochemistry and Plant Resources in West China (No. P2022-KF08), National Natural Science Foundation of China (Nos. 22107033 and U1703109), and the Scientific Research Project of Traditional Chinese Medicine of Hubei Provincial Health Commission (No. ZY2021M056). We thank the Analytical and Testing Center at Huazhong University of Science and Technology for single crystal X-ray diffraction data collection, and the Medical Subcenter of Huazhong University of Science and Technology for NMR data acquisition.
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Wang, Q. Shi, M. Dong, et al., Chem. Rev. 111 (2011) 76527709. doi: 10.1021/cr100147u
Y. Li, Y. Zhu, Z. Zhang, et al., Acta Pharm. Sin. B 10 (2020) 1073–1082. doi: 10.1016/j.apsb.2019.10.013
J. Zhou, Z. Zuo, J. Liu, et al., Org. Chem. Front. 7 (2020) 820828. doi: 10.1039/c9qo01538h
G.J. Zheng, P.F. Jin, L. Huang, et al., Bioorg. Chem. 99 (2020) 103794. doi: 10.1016/j.bioorg.2020.103794
N. Hamanaka, T. Matsumoto, Tetrahedron Lett. 13 (1972) 30873090. doi: 10.1016/S0040-4039(01)85015-2
S. Gasa, N. Hamanaka, S. Matsunaga, et al., Tetrahedron Lett. 17 (1976) 553556. doi: 10.1016/S0040-4039(00)77908-1
T. Kan, S. Hosokawa, S. Nara, et al., J. Org. Chem. 59 (1994) 55325534. doi: 10.1021/jo00098a009
K. Yu, Z. Yang, C. Liu, et al., Angew. Chem. Int. Ed. 58 (2019) 85568560. doi: 10.1002/anie.201903349
A. Turlik, Y. Chen, A.C. Scruse, et al., J. Am. Chem. Soc. 141 (2019) 80888092. doi: 10.1021/jacs.9b03751
L. Kong, H. Yu, M. Deng, et al., J. Am. Chem. Soc. 144 (2022) 52685273. doi: 10.1021/jacs.2c01692
J. Gao, P. Rao, K. Xu, et al., J. Am. Chem. Soc. 142 (2020) 45924597. doi: 10.1021/jacs.0c00308
Y. Kawamoto, F. Karube, T. Kobayashi, et al., Org. Lett. 22 (2020) 76097612. doi: 10.1021/acs.orglett.0c02811
Y. Kawamoto, F. Karube, T. Kobayashi, et al., Tetrahedron 83 (2021) 131958. doi: 10.1016/j.tet.2021.131958
S. Borrelly, L.A. Paquette, J. Am. Chem. Soc. 118 (1996) 727740. doi: 10.1021/ja9533454
S. Chow, C. Kress, N. Albaek, et al., Org. Lett. 13 (2011) 52865289. doi: 10.1021/ol202147r
C.W. Cunningham, W.M. Elballa, S.U. Vold, Neuropharmacology 151 (2019) 195207. doi: 10.1016/j.neuropharm.2019.03.006
P. Jin, G. Zheng, X. Yuan, et al., Bioorg. Chem. 111 (2021) 104870. doi: 10.1016/j.bioorg.2021.104870
D. Xin, P.J. Jones, N.C. Gonnella, J. Org. Chem. 83 (2018) 5035‒5043. doi: 10.1021/acs.joc.8b00338
N. Grimblat, M.M. Zanardi, A.M. Sarotti, J. Org. Chem. 80 (2015) 12526‒12534. doi: 10.1021/acs.joc.5b02396
S.G. Smith, J.M. Goodman, J. Am. Chem. Soc. 132 (2010) 129460112959.
R. Guo, T. Lv, F. Han, et al., Chin. Chem. Lett. 31 (2020) 12541258. doi: 10.1016/j.cclet.2019.09.042
J. Gao, X. Zhang, K. Shang, et al., Chin. Chem. Lett. 31 (2020) 427‒430. doi: 10.1016/j.cclet.2019.09.020
J. Zhou, J. Liu, T. Dang, et al., Org. Lett. 20 (2018) 20632066. doi: 10.1021/acs.orglett.8b00606
M. Jiang, Z. Wu, Q. Wu, et al., Chin. Chem. Lett. 32 (2021) 18931896. doi: 10.1016/j.cclet.2021.01.027
S. Parsons, H.D. Flack, T. Wagner, Acta Crystallogr. B 69 (2013) 249259. doi: 10.1107/S2052519213010014
S.F. El-Naggar, R.W. Doskotch, T.M. Odell, et al., J. Nat. Prod. 43 (1980) 617631. doi: 10.1021/np50011a016
M. Zhang, Y. Zhu, G. Zhan, et al., Org. Lett. 15 (2013) 30943097. doi: 10.1021/ol401292y
M. Katai, T. Terai, J. Katakawa, Mem. Osaka Inst. Technol., Ser. A 51 (2006) 16.
H. Zhang, H. Wang, L. Wang, et al., J. Asian Nat. Prod. Res. 7 (2005) 8790. doi: 10.1080/10286020310001609001
J.W. Burke, R.W. Doskotch, J. Nat. Prod. 53 (1990) 131137. doi: 10.1021/np50067a017
J. Wang, C.P. Zhang, P.K. Ouyang, Chem. Ind. Eng. Prog. 27 (2008) 702707.
H.S. Kang, J.H. Choi, W.K. Cho, et al., Arch. Pharm. Res. 27 (2004) 742750. doi: 10.1007/BF02980143
R. Mogana, K. Teng-Jin, C. Wiart, et al., Biomed. Res. Int. 2014 (2014) 903529.

Figure 1 Chemical structures of compounds 1−11 and the nomenclature of the unprecedented tetracyclic ring system in 1.

Figure 3 Linear correlation plots (A) and relative errors (B) between the experimental and calculated 13C NMR data for (5R*,8S*)- and (5S*,8R*)-1.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:






