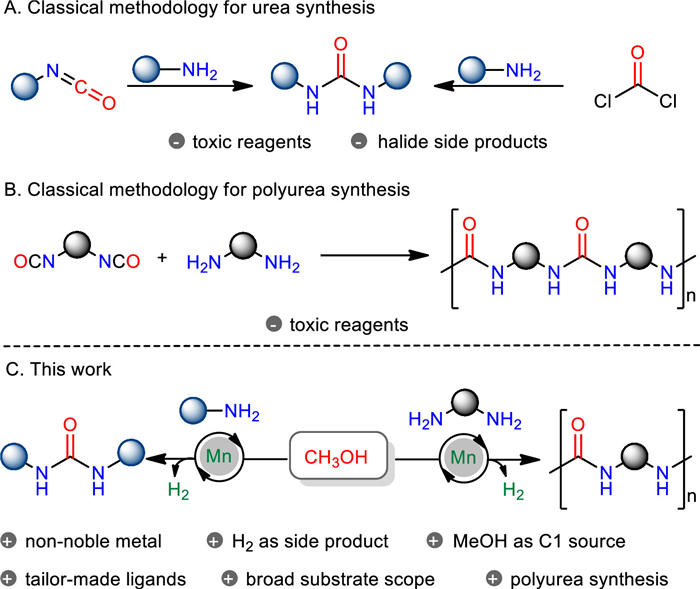
Scheme 1.
Synthesis of urea and polyurea.
Manganese catalyzed urea and polyurea synthesis using methanol as C1 source
Jiaxin Guo , Jun Tang , Hui Xi , Sheng-Yin Zhao , Weiping Liu

Ureas are a class of essential organic compounds and widely exist in pharmaceuticals, agrochemicals, dyes, antioxidants in gasoline as well as fine chemicals [1-4]. The corresponding polymers are usually found in substantial applications, such as coating, adhesive, materials, and others [5]. In the past decades, numerous approaches have been established for the synthesis of ureas and related polyureas [6]. Classical approaches toward the synthesis of (poly)ureas apply (di)isocyanates, phosgene, and its derivatives as starting materials (Scheme 1A) [7]. Although these processes proved to be very general, they suffer from serious toxicological and environmental issues. Alternatively, several methodologies have been established under (di)isocyanates-free conditions. For instance, the processes using carbon monoxide (CO) as the source of carbonyl fragment were developed for the ureas preparation [8-12]. Such carbonylation reactions are generally performed under high temperature and high CO pressure conditions with a stoichiometric amount of oxidants. Compared to CO, carbon dioxide (CO2) is more attractive because it is a renewable and non-toxic carbon resource [13-19]. However, this protocol typically requires high temperature and pressure, as well as dehydrating agents.
Recently, transition metal-based pincer catalysts [20-28] have showcased powerful catalytic ability in acceptorless dehydrogenative cross couplings with alcohols. Normally, H2O and/or H2 are generated as the "green" side products in the transformation [28]. This procedure potentially offers an attractive alternative for (poly)ureas synthesis using methanol to overcome the aforementioned problems, and such process avoids using toxic C1 source (such as isocyanates, phosgene, CO), oxidants, and dehydrating agents. Moreover, methanol is abundant, sustainable, biodegradable, and is also one of the most important C1 feedstock in organic synthesis [29].
In this regard, homogeneous catalysis enabled the dehydrogenative synthesis of urea using methanol as carbonyl fragment has been scarcely investigated. In 2016, Hong group demonstrated the feasibility of this transformation by utilizing methanol and amines in the presence of ruthenium complex as the catalyst [30]. Recently, the sole dehydrogenative preparation of polyureas utilizing diamines and methanol was realized by Kumar group applying ruthenium catalysis [31]. However, the catalyst based on precious ruthenium is less desirable compared to non-noble metals, due to its less abundant and more expensive nature. Moreover, Hazar/Bernskoetter and coworkers elegantly developed iron-catalyzed urea synthesis with methanol and amines. In this case, a large excess amount of amines were required to afford high yields and TONs [32], and demonstrated poor substrate scope.
Nowadays, a rising trend in catalysis is the application of 3d metals due to their natural abundance and often reduced toxicity [22-26, 33-42]. Particularly, manganese is overall the twelfth most abundant element and the third most abundant transition metal in the earth's crust. Furthermore, manganese is found as an essential trace element for life [43]. In recent years, the well-defined pincer manganese catalysts have been well explored [44-53]. Herein, we demonstrate the manganese-catalyzed urea and polyurea synthesis using methanol as carbonyl source. The presence of newly synthesized MACHO ligands permits increased efficiency/activity using amines and methanol under mild conditions, thereby providing the ureas and polyureas with broad substrate scope in good to excellent yields.
We initiated our investigations by testing the dehydrogenative coupling between benzylamine (1a) and methanol as a benchmark reaction in the presence of Mn-MACHO catalyst. Firstly, different solvents were screened with a catalytic amount of KOtBu as the base, toluene was proved slightly better than mesitylene, 1, 4-dioxane, and xylene (Table 1, entries 1–5), affording the desired product 1, 3-dibenzylurea (2a) in 86% yield. Other tested bases, such as KH, NaOtBu and KOMe, provided comparable efficiency (Table 1, entries 6–8). Surprisingly, decreasing the substrate concentration (2.0 mol/L) led to a much-diminished yield (49%) of 2a (entry 9), while a higher concentration (10 mol/L) did not affect much thus delivering the corresponding urea 2a in 85% yield (entry 10). Unfortunately, lower yields were obtained when the reactions were carried out under milder conditions (Table 1, entries 11 and 12).
Recently, Mn-MACHOiPr catalyst (Mn-2) has displayed high activities in hydrogenation and dehydrogenation processes [44-47, 54-57]. In this regard, Mn-2 exhibited lower activity than Mn-1 in the current transformation under 130 ℃ conditions (Table 2, entries 1 and 2). Prompted by the critical role of substituents on phosphorus, we became interested in the synthesis of Mn-MACHO catalysts with different substitutions on the aromatic rings to improve efficiency (Table 2). Four new Mn-MACHO catalysts as shown in Table 2 were successfully synthesized. Suitable crystals of complex Mn-3, Mn-4 and Mn-6 for X-ray diffraction (XRD) measurement were grown via recrystallization from a mixture of dichloromethane (DCM) and n-hexane solution (see Supporting information). Unfortunately, Mn-5 failed to get suitable crystals for XRD measurement, but it was fully characterized by NMR and HRMS analyses. The catalytic activities were further tested in the current system under milder conditions (130 ℃). We found that a comparable yield of 2a was obtained when phenyl groups of MACHO ligand were replaced by 2-naphthyl group (Mn-3) at the phosphorus center (Table 2, entry 3). To our delight, while a methoxy (Mn-4) or methyl (Mn-5) group was installed at para-position of the phenyl ring, catalysts exhibited higher activities, thus providing the desired product in 74% and 76% yields, respectively (Table 2, entries 4 and 5). The yield of 2a was slightly improved up to 84% by applying the sterically bulky 3, 5-dimethyl group substituted in the phenyl ring of MACHO ligand (Mn-6) (Table 2, entry 6). Delightedly, increasing the loading amount of methanol led to 91% yield of 2a (Table 2, entry 6). Furthermore, it was observed that the TON of the current system could achieve 152 with lower catalyst loading for the product 1a, which was comparable to reported iron system, but lower than ruthenium catalysis.
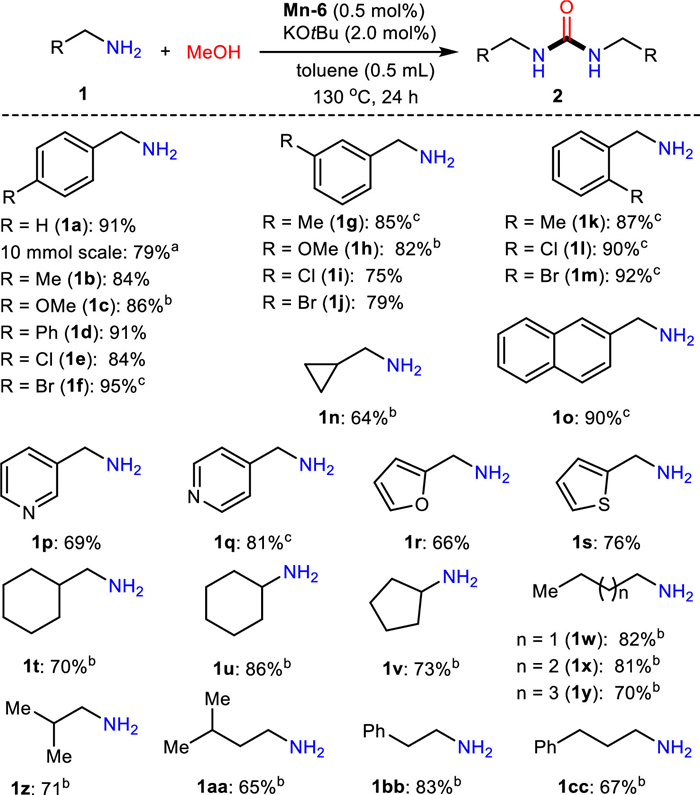
Encouraged by the success, we explored the substrate scope under the optimized catalyst (Mn-6) and conditions. As shown in Scheme 2, the desired products were obtained in all cases with moderate to excellent yields. Various substituents and functional groups on the benzene ring, including methoxy-, chloro-, bromo- and others, irrespective of their location at the ortho-, meta- or para-position could be well tolerated. It should be pointed out that this transformation could not tolerate strong electron-withdrawing substituents, for example, trifluoromethyl and nitro groups. Notably, heteroaryl substituted methylamines, such as pyridyl (1p, 1q), furanyl (1r), and thienyl (1s), could deliver the corresponding ureas smoothly as well under the manganese catalysis. It was found that a series of aliphatic amines were also suitable substrates, thus affording the desired urea products in good yields under a slightly higher amount of catalyst loading conditions. However, secondary amines, such as N-methylbenzylamine, failed to furnish the desired urea products under the developed manganese catalyst system, along with the formation of formamide as the major product. Additionally, we also tested the tolerance of substrates bearing other reducible functional groups. These experiments were conducted with 1.0 equiv. of additional substrates (Table S1 in Supporting information). We observed 1, 3-dibenzylurea (2a) was formed in moderate to high yields in the presence of acetophenone and 1, 2-diphenylethyne, albeit along with hydrogenation to the corresponding alcohol and alkene occurred. Moreover, the reactions were stopped upon the addition of primary amide substrates. Importantly, a slightly lower yield of 2a was obtained when performing the manganese-catalyzed dehydrogenative coupling on a larger 10 mmol scale.
Delightedly, the applied PNP-based manganese catalyzed dehydrogenative coupling system was not restricted to the synthesis of normal urea. Indeed, diamines also underwent the process with methanol for the formation of corresponding polyureas with high catalytic efficacy albeit with moderate to good molecular weight (Mn) (Table 3). More specifically, a set of conditions were tested using 4, 7, 10-trioxa-1, 13-tridecanediamine (3a) as the substrate, the product was afforded with 86% yield and 2752 Da, employing the combination of Mn-6 (0.5 mol%) and KOtBu (2.0 mol%) under 150 ℃ (Table S2 in Supplementary Material). Other diamines, such as hexane-1, 6-diamine (3b), octane-1, 8-diamine (3c), and 4, 4′-methylenebis(cyclohexan-1-amine) (3d), provided the corresponding polyureas as well using a mixed solvent system due to poor solubility of the polymers in toluene.
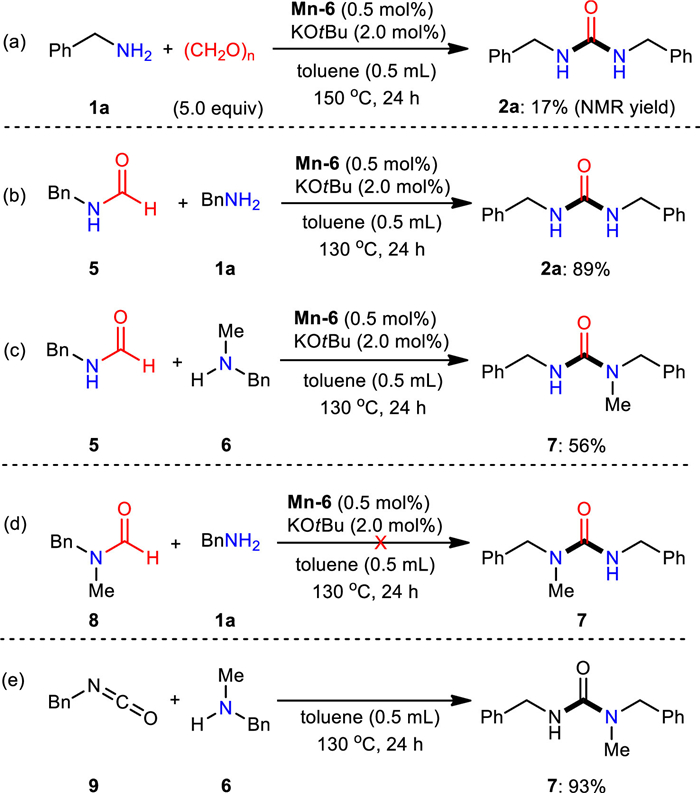
To better understand the performance of this manganese catalyzed dehydrogenative transformation, a set of mechanistic experiments were carried out (Scheme 3). Employing paraformaldehyde instead of methanol led to the formation of 2a in 17% NMR yield (Scheme 3a). This result demonstrated that formaldehyde is a potential reaction intermediate in the dehydrogenative cross coupling process [58]. However, the diminished productive yield was probably due to the low propensity for the formation of [Mn]H2 species in the presence of paraformaldehyde [59, 60]. The experiment using N-benzylformamide (5) and benzylamine (1a) or N-methyl-1-phenylmethanamine (6) as substrates under the standard condition was conducted [61, 62], the desired 1, 3-dibenzylurea (2a) and 1, 3-dibenzyl-1-methylurea (7) resulted in 89% and 56% yields, respectively (Schemes 3b and c). Additionally, applying N-benzyl-N-methylformamide (8) along with benzylamine (1a) as starting materials failed to form the corresponding urea product (Scheme 3d), which is also in agreement with the formation of formylated [58, 63-67] product using N-methylbenzylamine as substrate (vide supra). These findings highlight the importance of the free NH moiety of formamide [68]. Furthermore, reaction applying 9 and 6 was executed, and an excellent yield of desired urea 7 was obtained (Scheme 3e).
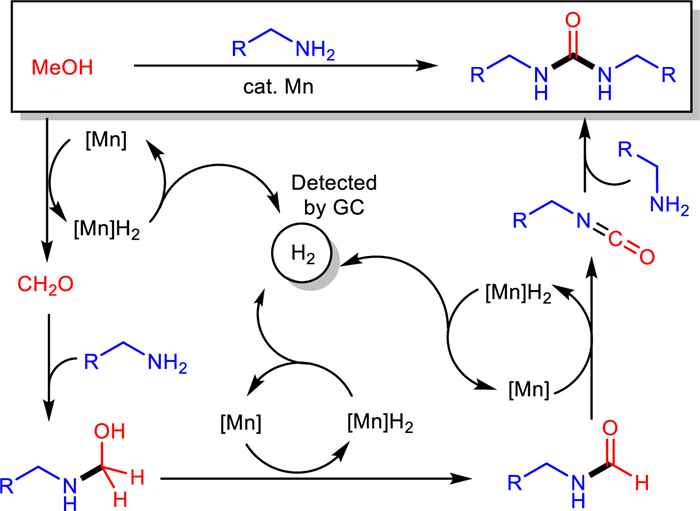
Based on the above observations, we postulate NH-formamide and isocyanate as the key intermediates in the pathway of this dehydrogenative reaction. Here, plausible reaction pathways for the manganese-catalyzed urea synthesis with methanol as carbonyl source are proposed (Scheme 4). Formaldehyde is generated firstly via dehydrogenation of methanol under manganese catalysis, followed by reacting with the amine to afford hemiaminal species. Subsequent elimination of H2 furnishes formamide as one of the key intermediates. Then, formamide is dehydrogenated enabled by the manganese catalyst to form a transient isocyanate, which is attacked by another equivalent of amine releasing the final product. Notably, the formation of H2 was confirmed by GC via analyzing the gas existent in the headspace of the reaction tube (see Supporting information).
In summary, manganese-catalyzed urea synthesis via dehydrogenative coupling of methanol and amines has been established for the first time. The presented newly synthesized MACHO ligands demonstrations increased efficiency/activity under mild conditions, thereby providing the ureas with broad substrate scope in good to excellent yields. Additionally, diamines can be successfully applied in this protocol for access to the corresponding polyureas. Preliminary mechanistic investigations reveal that in situ formed NH-formamide and isocyanate are the central intermediates in this dehydrogenative transformation.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This article is dedicated to Professor Matthias Beller on the occasion of his 60th birthday. The authors thank Prof. Dr Weimin Xuan (Donghua University) for his great support with the X-ray diffraction analysis. We also thank the financial support from the Fundamental Research Funds for the Central Universities (No. 2232022A-09), the National Natural Science Foundation of China (No. 22001033), Natural Science Foundation of Shanghai (No. 20ZR1401100).
Supplementary material associated with this article can be found, in the online version, at doi:
B. Kocyigit-Kaymakcioglu, A.O. Celen, N. Tabanca, et al., Molecules 18 (2013) 3562–3576. doi: 10.3390/molecules18033562
C. Nitschke, G. Scherr, Ullmann's Encyclopedia of Industrial Chemistry, WileyVCH, Weinheim, 2010.
K. Matsuda, Med. Res. Rev. 14 (1994) 271–305. doi: 10.1002/med.2610140302
D. Bankston, J. Dumas, R. Natero, et al., Org. Proc. Res. Dev. 6 (2002) 777–781. doi: 10.1021/op020205n
G.A. Howarth, Surf. Coat. Int. Pt. B-Coat. Trans. 86 (2003) 111–118. doi: 10.1007/BF02699621
F. Bigi, R. Maggi, G. Sartori, Green Chem. 2 (2000) 140–148. doi: 10.1039/b002127j
H. Babad, A.G. Zeiler, Chem. Rev. 73 (1973) 75–91. doi: 10.1021/cr60281a005
Y. Cao, J.G. Yang, Y. Deng, et al., Angew. Chem. Int. Ed. 59 (2020) 2080–2084. doi: 10.1002/anie.201914089
E.S. Smirnova, J.M. Muñoz Molina, A. Johnson, et al., Angew. Chem. Int. Ed. 55 (2016) 7487–7491. doi: 10.1002/anie.201603200
Z.H. Guan, H. Lei, M. Chen, et al., Adv. Synth. Catal. 354 (2012) 489–496. doi: 10.1002/adsc.201100545
J.H. Park, J.C. Yoon, Y.K. Chung, Adv. Synth. Catal. 351 (2009) 1233–1237. doi: 10.1002/adsc.200900106
K. Orito, M. Miyazawa, T. Nakamura, et al., J. Org. Chem. 71 (2006) 5951–5958. doi: 10.1021/jo060612n
Y. Zhao, X. Guo, Z. Si, et al., J. Org. Chem. 85 (2020) 13347–13353. doi: 10.1021/acs.joc.0c02032
S. Jiang, H.Y. Cheng, R.H. Shi, et al., ACS Appl. Mater. Interfaces 11 (2019) 47413–47421. doi: 10.1021/acsami.9b17677
P. Wu, H. Cheng, R. Shi, et al., Adv. Synth. Catal. 361 (2019) 317–325. doi: 10.1002/adsc.201801134
S. Jiang, R. Shi, H. Cheng, C. Zhang, F. Zhao, Green Energy Environ. 2 (2017) 370–376. doi: 10.1016/j.gee.2017.05.001
P. Wang, Y. Fei, Y. Long, Y. Deng, J. CO2 Util. 28 (2018) 403–407. doi: 10.1016/j.jcou.2018.10.020
M. Xu, A.R. Jupp, D.W. Stephan, Angew. Chem. Int. Ed. 56 (2017) 14277–14281. doi: 10.1002/anie.201708921
M. Tamura, K. Ito, Y. Nakagawa, K. Tomishige, J. Catal. 343 (2016) 75–85. doi: 10.1016/j.jcat.2015.11.015
B.G. Reed-Berendt, D.E. Latham, M.B. Dambatta, L.C. Morrill, ACS Cent. Sci. 7 (2021) 570–585. doi: 10.1021/acscentsci.1c00125
D.A. Ekanayake, H. Guan, Top. Organomet. Chem. 68 (2021) 263–320.
L. Alig, M. Fritz, S. Schneider, Chem. Rev. 119 (2019) 2681–2751. doi: 10.1021/acs.chemrev.8b00555
K. Junge, V. Papa, M. Beller, Chem. Eur. J. 25 (2019) 122–143. doi: 10.1002/chem.201803016
A. Mukherjee, D. Milstein, ACS Catal. 8 (2018) 11435–11469. doi: 10.1021/acscatal.8b02869
S. Chakraborty, P. Bhattacharya, H. Dai, H. Guan, Acc. Chem. Res. 48 (2015) 1995–2003. doi: 10.1021/acs.accounts.5b00055
T. Zell, D. Milstein, Acc. Chem. Res. 48 (2015) 1979–1994. doi: 10.1021/acs.accounts.5b00027
C. Gunanathan, D. Milstein, Chem. Rev. 114 (2014) 12024–12087. doi: 10.1021/cr5002782
C. Gunanathan, D. Milstein, Science 341 (2013) 1229712–1229723. doi: 10.1126/science.1229712
K. Natte, H. Neumann, M. Beller, R.V. Jagadeesh, Angew. Chem. Int. Ed. 56 (2017) 6384–6394. doi: 10.1002/anie.201612520
S.H. Kim, S.H. Hong, Org. Lett. 18 (2016) 212–215. doi: 10.1021/acs.orglett.5b03328
A. Kumar, D. Armstrong, G. Peters, M. Nagala, S. Shirran, Chem. Commun. 57 (2021) 6153–6156. doi: 10.1039/D1CC01121A
E.M. Lane, N. Hazari, W.H. Bernskoetter, Chem. Sci. 9 (2018) 4003–4008. doi: 10.1039/C8SC00775F
J. Wen, F. Wang, X. Zhang, Chem. Soc. Rev. 50 (2021) 3211–3237. doi: 10.1039/D0CS00082E
W. Ai, R. Zhong, X. Liu, Q. Liu, Chem. Rev. 119 (2019) 2876–2953. doi: 10.1021/acs.chemrev.8b00404
W. Liu, B. Sahoo, K. Junge, M. Beller, Acc. Chem. Res. 51 (2018) 1858–1869. doi: 10.1021/acs.accounts.8b00262
F. Kallmeier, R. Kempe, Angew. Chem. Int. Ed. 57 (2018) 46–60. doi: 10.1002/anie.201709010
G.A. Filonenko, R. van Putten, E.J.M. Hensen, E.A. Pidko, Chem. Soc. Rev. 47 (2018) 1459–1483. doi: 10.1039/C7CS00334J
J. Chen, J. Guo, Z. Lu, Chin. J. Chem. 36 (2018) 1075–1109. doi: 10.1002/cjoc.201800314
X. Du, Z. Huang, ACS Catal. 7 (2017) 1227–1243. doi: 10.1021/acscatal.6b02990
P. Chirik, R. Morris, Acc. Chem. Res. 48 (2015) 2495 -2495. doi: 10.1021/acs.accounts.5b00385
R.M. Bullock, Science 342 (2013) 1054–1055. doi: 10.1126/science.1247240
Z. Yan, P.L. Shao, Q. Qiang, et al., Chin. Chem. Lett. 33 (2022) 1207–1226. doi: 10.1016/j.cclet.2021.08.112
D.S. Avila, R.L. Puntel, M. Aschner, Manganese in health and disease, in: A. Sigel, H. Sigel, K.O.R. Sigel (Eds.), Interrelations Between Essential Metal Ions and Human Diseases, Springer, Netherlands, Dordrecht, 2013, pp. 199–227.
S. Elangovan, C. Topf, S. Fischer, et al., J. Am. Chem. Soc. 138 (2016) 8809–8814. doi: 10.1021/jacs.6b03709
S. Fu, Z. Shao, Y. Wang, Q. Liu, J. Am. Chem. Soc. 139 (2017) 11941–11948. doi: 10.1021/jacs.7b05939
A. Kaithal, P. van Bonn, M. Hölscher, W. Leitner, Angew. Chem. Int. Ed. 59 (2020) 215–220. doi: 10.1002/anie.201909035
P. Ryabchuk, K. Stier, K. Junge, M.P. Checinski, M. Beller, J. Am. Chem. Soc. 141 (2019) 16923–16929. doi: 10.1021/jacs.9b08990
F. Kallmeier, T. Irrgang, T. Dietel, R. Kempe, Angew. Chem. Int. Ed. 55 (2016) 11806–11809. doi: 10.1002/anie.201606218
M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier, K. Kirchner, J. Am. Chem. Soc. 138 (2016) 15543–15546. doi: 10.1021/jacs.6b10433
L. Zhang, Z. Wang, Z. Han, K. Ding, Angew. Chem. Int. Ed. 59 (2020) 15565–15569. doi: 10.1002/anie.202006383
J. Sklyaruk, J.C. Borghs, O. El-Sepelgy, M. Rueping, Angew. Chem. Int. Ed. 58 (2019) 775–779. doi: 10.1002/anie.201810885
A. Mukherjee, A. Nerush, G. Leitus, et al., J. Am. Chem. Soc. 138 (2016) 4298–4301. doi: 10.1021/jacs.5b13519
C. Huang, J. Liu, H.H. Huang, X. Xu, Z. Ke, Chin. Chem. Lett. 33 (2022) 262–265. doi: 10.1016/j.cclet.2021.06.046
A. Kaithal, M. Hölscher, W. Leitner, Angew. Chem. Int. Ed. 57 (2018) 13449–13453. doi: 10.1002/anie.201808676
S. Kar, A. Goeppert, J. Kothandaraman, G.K.S. Prakash, ACS Catal. 7 (2017) 6347–6351. doi: 10.1021/acscatal.7b02066
X. Liu, T. Werner, Adv. Synth. Catal. 363 (2021) 1096–1104. doi: 10.1002/adsc.202001209
B.G. Reed-Berendt, L.C. Morrill, J. Org. Chem. 84 (2019) 3715–3724. doi: 10.1021/acs.joc.9b00203
Z. Shao, Y. Li, C. Liu, et al., Nat. Commun. 11 (2020) 591–598. doi: 10.1038/s41467-020-14380-3
L.U. Nordstrøm, H. Vogt, R. Madsen, J. Am. Chem. Soc. 130 (2008) 17672–17673. doi: 10.1021/ja808129p
Y. Zhang, C. Chen, S.C. Ghosh, Y. Li, S.H. Hong, Organometallics 29 (2010) 1374–1378. doi: 10.1021/om901020h
S. Kotachi, Y. Tsuji, T. Kondo, Y. Watanabe, ChemInform 21 (1990) 549–550. doi: 10.1002/chin.199041152
G.S. Kumar, R.A. Kumar, P.S. Kumar, et al., Chem. Commun. 49 (2013) 6686–6688. doi: 10.1039/c3cc42381f
S. Chakraborty, U. Gellrich, Y. Diskin-Posner, et al., Angew. Chem. Int. Ed. 56 (2017) 4229–4233. doi: 10.1002/anie.201700681
G. Choi, S.H. Hong, ACS Sustain. Chem. Eng. 7 (2018) 716–723.
B. Kang, S.H. Hong, Adv. Synth. Catal. 357 (2015) 834–840. doi: 10.1002/adsc.201400982
N. Ortega, C. Richter, F. Glorius, Org. Lett. 15 (2013) 1776–1779. doi: 10.1021/ol400639m
E.M. Lane, K.B. Uttley, N. Hazari, W. Bernskoetter, Organometallics 36 (2017) 2020–2025. doi: 10.1021/acs.organomet.7b00258
J. Bruffaerts, N. von Wolff, Y. Diskin-Posner, Y. Ben-David, D. Milstein, J. Am. Chem. Soc. 141 (2019) 16486–16493. doi: 10.1021/jacs.9b08942

Scheme 2 Manganese-catalyzed urea synthesis. Reaction conditions: 1 (2.0 mmol, 1.0 equiv.), MeOH (3.0 mmol, 1.5 equiv.), Mn-6 (0.01 mmol, 0.5 mol%), KOtBu (2.0 mol%) and toluene (0.5 mL) in 50 mL pressure tube, 130 ℃, isolated yields. a 1a (10 mmol) and MeOH (15 mmol) in 250 mL pressure tube, 150 ℃. b Mn-6 (0.75 mol%), KOtBu (3.0 mol%). c 150 ℃.

Scheme 4 Plausible reaction pathways for the dehydrogenative cross coupling of methanol and amines.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:




 下载:
下载:
