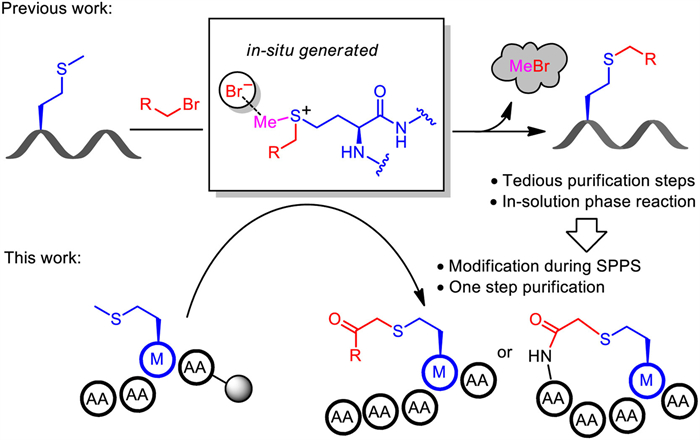
Figure 1.
One-pot demethylative alkylation of Met residue.
On-resin peptide modification of methionine residue by employing 2-bromoacetate derivatives
Qi-Long Hu , Jia-Tian Liu , Guangjin Fan , Jian Li , Yixian Li , Albert S.C. Chan , Xiao-Feng Xiong

Peptides and peptidomimetics are important classes of bioactive compounds in natural products, which profoundly impact the modern pharmaceutical industry [1]. However, such peptides are mainly obtained via isolation or de novo synthesis [2,3]. Taking advantage of solid-phase peptide synthesis (SPPS), the surge of synthetic peptides has broadened the sources of bioactive agents and improved the rapid development of peptide science [4-6]. Modified peptides often feature improved pharmacokinetics, bioavailability, and proteolytic stability, which are considered to be excellent alternatives compared to small molecules [7-10]. Therefore, developing chemical methods to achieve versatile peptide modification is desirable.
On-resin modification during the SPPS presents a step-economic approach to obtain modified peptides without tedious purification procedures and provides an indirect approach to alleviate the requirements of functional group tolerance [11-14]. However, the reported methods relied on the subtle designed orthogonal protection strategies or the extra preloaded reaction sites, which may limit their applications [15-18]. The redox-sensitive and nucleophilic thioether sidechain of methionine (Met) could be alkylated even under acidic reaction conditions, making it an ideal handle to achieve peptide modification [19,20]. Upon this unique residue, bioconjugation could be realized via the formation of sulfonium, sulfilimine, and newly generated thermodynamically stable thioether [21-27]. Although few strategies have been explored in this realm, the employment of Met residue to construct cyclic peptides is rarely reported.
Recently, our group reported the demethylative alkylation of Met by employing sulfonium as the key intermediate (Fig. 1), which demonstrated an efficient method to achieve sidechain diversity of the relevant peptides in solution [28]. However, tedious purification procedures are required to obtain the desired products, and the functional group tolerance might be problematic. Considering the carboxylic functional group is an ideal C1 carbon linkage for peptide modification and macrocyclization, and SPPS is an efficient strategy that could avoid the tedious purification steps, we envisioned whether the on-resin peptide modification and macrocyclization could be achieved by the demethylative alkylation on Met residue. Herein, we advanced the strategy for Met functionalization by combining the advantages of SPPS and one-pot proceeding modification. The key intermediate was in-situ generated on resin to afford the modified product or cyclic peptide, which improved the efficiency and expanded the potential utilities of this method in medicinal chemistry.
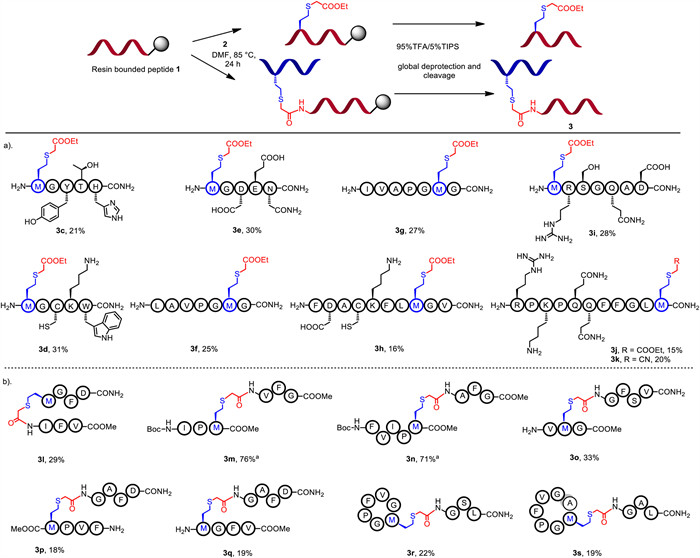
We initiated our study by preparing the model peptide 1a bonded on rink amide resin (0.64 mmol/g) with 0.1 mmol scales and employing 2.0 equiv. of ethyl 2-bromoacetate 2a as the modification reagent. To avoid the tedious purification procedures, the reaction was carried out under 85 ℃ in DMF and then purified after global deprotection and cleavage to afford the final product (Figs. S3-S16 in Supporting information for details). To our delight, desired product 3a was obtained in 37% isolated yield calculated from resin loading (Table 1, entry 1). We also investigated this method by employing 2-chlorotrityl chloride (2-CTC) and Wang resin, which were frequently used to prepare peptides with carboxylated C-terminus. Peptide bounded on Wang resin was successfully decorated to afford 3b in 17% overall yield (Table 1, entry 2). The 2-CTC resin was sensitive to high temperature, resulting in the cleavage of peptide and trace amount of 3b being detected in the cleavage solution. However, N-Boc protected 3b was isolated from reaction solution in 26% yield (Table 1, entry 3). Employment of MeCN as reaction solvent or adding 4.0 equiv. of 2a, the yield of 3a could be maintained (Table 1, entries 4 and 5). Similar results were also observed after employing the rink amide resin with different loadings, delivering 3a in the yields of 34%−36% (Table 1, entries 6 and 7).
With the optimized conditions in hand, we directed our effort towards the on-resin peptide modification (Fig. 2a). Peptide (5-mer to 10-mer) substrates prepared by standard Fmoc-SPPS were subjected to direct modification by 2a before the global deprotection and cleavage. Excitingly, peptides containing amino acid variants including tryptophan (Trp), tyrosine (Tyr), lysine (Lys), aspartic acid (Asp), histidine (His), arginine (Arg) or cysteine (Cys), all successfully converted to the corresponding modified products 3c-3h with the total yields of 16%−31% including peptide synthesis steps, highlighting the step-economy of this protocol. Moreover, modification of substance P (SP) and microcin C7 (McC7) could be achieved via this strategy, affording the desired products 3i-3k with comprehensive yields. Next, 2-bromoacetyl modification reagents derived from peptides were also investigated (Fig. S1 in Supporting information), which could be developed as a potential fragment condensation approach without the addition of an extra coupling reagent [29]. Peptide 2c with a 2-bromoacetyl cap on N-terminus was synthesized and evaluated, the desired product 3l was obtained in 29% yield. To illustrate the efficiency of this crucial step, two relevant peptide fragments were prepared respectively (Fig. S1) and reacted in MeCN to afford the linked products 3m and 3n in more than 70% yields. We then optimized this strategy by developing an on-resin acylation protocol, which could be further applied to construct cyclic peptides. The results of peptide-peptide fragment condensation were encouraging, as the 2-bromoacetyl linker turned out to be an ideal linkage between two different peptide fragments, affording the corresponding products 3o-3s in 18%−33% yields (Fig. 2b).
Based on these robust results, we challenged our method to construct cyclic peptides by combining on-resin acylation and macrocyclization [30-33]. Resin-bonded peptide precursors containing 2-bromoacetyl cap on N-terminus were prepared for investigation, including (i, i + 2) to (i, i + 8) cyclization sites. The reactions were carried out under 85 ℃ in DMF (0.2 mmol/25 mL), and cyclic peptides as final products were obtained via one-step purification with the overall yields of 7%−28% calculated from resin loading (Fig. 3). Cyclization at the (i, i + 2) position was failed, probably due to the unfavored ring strain (Fig. S75 in Supporting information). To our delight, the highest conversion was observed for (i, i + 4) macrocyclization product, either elongating or truncating the peptide sequence led to decreasing yields. Typically, diastereoisomers were observed for (i, i + 3), (i, i + 5), and (i, i + 7) cyclopeptides in DMSO-d6, but did not appear for (i, i + 4), (i, i + 6), and (i, i + 8) products. Notably, the distinctive product 5d bearing a d-valine (Val) was cyclized at the (i, i + 3) position, but no dr value was observed in NMR spectrum. We speculated that the induction of a d-Val in N-terminus was necessary to reverse the contrary cyclization orientation avoiding the flipping process, which resulted in the disappearance of diastereoisomers [35,36]. The (i, i+5) cyclic peptide 5g and its counterpart 5g’ bearing a d-Val l were synthesized to explain the results. NMR analysis showed 3:1 dr values of both peptides, which demonstrated the induction of a d-configuration residue in the N-terminus could help to reduce the dr value and proved our hypothesis. Furthermore, the dr value of 5g converted to 5:1 when characterizing in MeOD-d4, indicating the dr value might be resulted from the configuration shifting of cyclic peptide (for details see Fig. S155 in Supporting information).
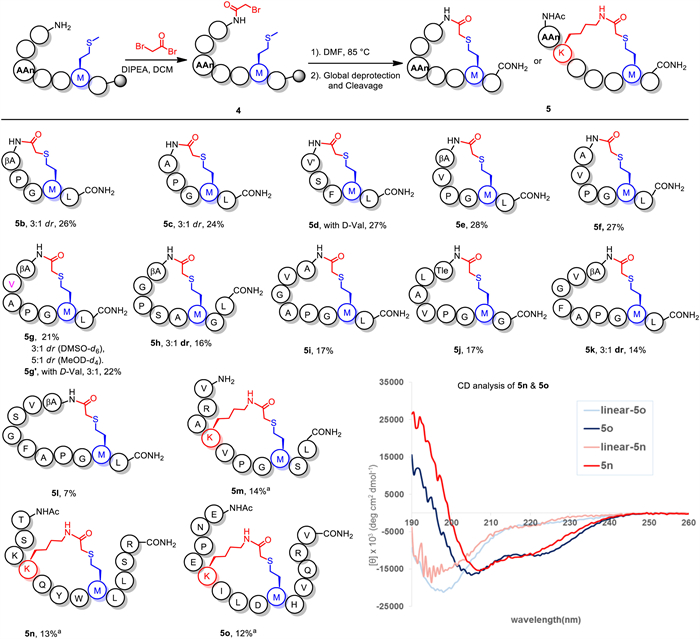
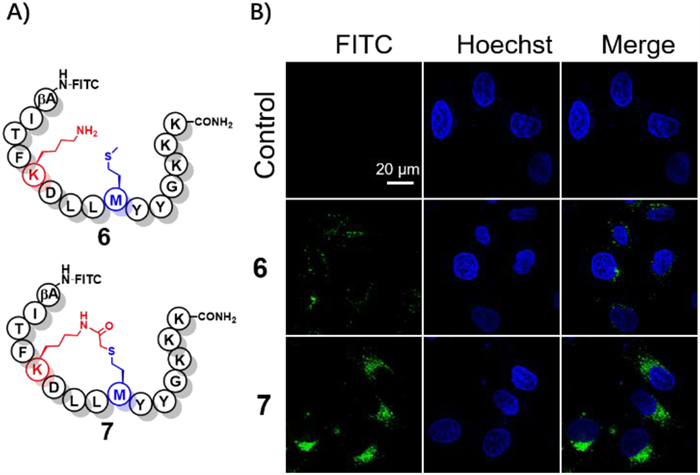
Stapled peptides containing a preformed and stable α-helical conformation, exhibited improved membrane permeability compared to linear bioactive peptides [31,34,37]. To expand the utility of this method in constructing helix-constrained peptides four stapled peptides were synthesized with the 13% average yields from resin loading (5m-5o and 7). Circular dichroism (CD) analysis indicated the α-helical structure of 5n and 5o in an aqueous solution (about 20% helicity) [38], suggesting that cross-linking of Met-Lys sidechains resulted in improved helicity when attached to their linear precursors (Table S1 in Supporting information). To further investigate the cellular uptake properties of stapled peptide 7 and its linear precursor 6 in HeLa cells, fluorescein isothiocyanate (FITC) was introduced on N-terminus (Fig. 4). The results of confocal microscopy imaging indicated that peptide 7 displayed diffuse and enhanced cellular uptake rates compared to its linear precursor, demonstrating the practical utilities of this method in medicinal chemistry.
In conclusion, we developed an easy-to-operate approach to achieve Met residue functionalization by employing 2-bromoacetate derivatives, which combined the advantages of SPPS and the efficiency of one-pot proceeding modification. Moreover, our method could be applied to prepare the modified natural product and peptide cycles of different sizes, demonstrating its practical utilities and potential for medicinal chemistry research.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors acknowledge financial supported by the National Natural Science Foundation of China (No. 22077144), Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2018B030306017), Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery (No. 2019B030301005), Key Research and Development Program of Guangdong Province (No. 2020B1111110003).
Supplementary material associated with this article can be found, in the online version, at doi:
A. Henninot, J.C. Collins, J.M. Nuss, J. Med. Chem. 61 (2018) 1382–1414. doi: 10.1021/acs.jmedchem.7b00318
U. Kazmaier, L. Junk, Mar. Drugs 19 (2021) 446. doi: 10.3390/md19080446
A.T. Bockus, C.M. McEwen, R.S. Lokey, Curr. Top. Med. Chem. 13 (2013) 821–836. doi: 10.2174/1568026611313070005
J.M. Palomo, RSC Adv. 4 (2014) 32658–32672. doi: 10.1039/C4RA02458C
J.L. Lau, M.K. Dunn, Bioorg. Med. Chem. 26 (2018) 2700–2707. doi: 10.1016/j.bmc.2017.06.052
E. Lenci, A. Trabocchi, Chem. Soc. Rev. 49 (2020) 3262–3277. doi: 10.1039/d0cs00102c
G.J.B. Philippe, D.J. Craik, S.T. Henriques, Drug Discov. Today 26 (2021) 1521–1531. doi: 10.1016/j.drudis.2021.01.022
A.D. Cunningham, N. Qvit, D. Mochly-Rosen, Curr. Opin. Struc. Biol. 44 (2017) 59–66.
B.M. Cooper, J. Iegre, D. O’ Donovan, M.O. Halvarsson, D.R. Spring, Chem. Soc. Rev. 50 (2021) 1480–1494. doi: 10.1039/d0cs00556h
P. Servatius, L. Junk, U. Kazmaier, Synlett 30 (2019) 1289–1302. doi: 10.1055/s-0037-1612417
H. Shen, X. Ning, D. Liu, S. Liu, M. Zhang, Synlett 29 (2018) 2588–2594. doi: 10.1055/s-0037-1611060
N. Cankařová, E. Schütznerová, V. Krchňák, Chem. Rev. 119 (2019) 12089–12207. doi: 10.1021/acs.chemrev.9b00465
Y. Zhou, X.W. Liang, RSC Adv. 11 (2021) 37942–37951. doi: 10.1039/d1ra07503a
H. Itoh, M. Inoue, Org. Biomol. Chem. 17 (2019) 6519–6527. doi: 10.1039/c9ob01130g
L. Reguera, D.G. Rivera, Chem. Rev. 119 (2019) 9836–9860. doi: 10.1021/acs.chemrev.8b00744
V. Sarojini, A.J. Cameron, K.G. Varnava, W.A. Denny, G. Sanjayan, Chem. Rev. 119 (2019) 10318–10359. doi: 10.1021/acs.chemrev.8b00737
M.C. Morejon, A. Laub, B. Westermann, D.G. Rivera, L.A. Wessjohann, Org. Lett. 18 (2016) 4096–4099. doi: 10.1021/acs.orglett.6b02001
W.H. So, Y. Bao, X. Chen, J. Xia, Org. Lett. 23 (2021) 8277–8281. doi: 10.1021/acs.orglett.1c03014
J. Zang, Y.L. Chen, W.X. Zhu, S.X. Lin, Biochemistry 59 (2020) 132–138. doi: 10.1021/acs.biochem.9b00789
J.N. deGruyter, L.R. Malins, P.S. Baran, Biochemistry 56 (2017) 3863–3873. doi: 10.1021/acs.biochem.7b00536
S. Lin, X. Yang, S. Jia, et al., Science 355 (2017) 597–602. doi: 10.1126/science.aal3316
M.T. Taylor, J.E. Nelson, M.G. Suero, M.J. Gaunt, Nature 562 (2018) 563–568. doi: 10.1038/s41586-018-0608-y
J. Kim, B.X. Li, R.Y. Huang, et al., J. Am. Chem. Soc. 142 (2020) 21260–21266. doi: 10.1021/jacs.0c09926
W.A. Scott, E.G. Gharakhanian, A.G. Bell, et al., J. Am. Chem. Soc. 143 (2021) 18196–18203. doi: 10.1021/jacs.1c07925
D. Lin, M. Wallace, A.J. Allentoff, et al., Bioconjugate Chem. 31 (2020) 1908–1916. doi: 10.1021/acs.bioconjchem.0c00256
L.M.B. Anaya, R. Petitdemange, M. Rosselin, et al., Biomacromolecules 22 (2021) 76–85. doi: 10.1021/acs.biomac.0c00374
R. Petitdemange, E. Garanger, L. Bataille, et al., Biomacromolecules 18 (2017) 544–550. doi: 10.1021/acs.biomac.6b01696
Q.L. Hu, J.T. Liu, J. Li, et al., Org. Lett. 23 (2021) 8543–8548. doi: 10.1021/acs.orglett.1c03241
H.Y. Chow, Y. Zhang, E. Matheson, X. Li, Chem. Rev. 119 (2019) 9971–10001. doi: 10.1021/acs.chemrev.8b00657
R. Jwad, D. Weissberger, L. Hunter, Chem. Rev. 120 (2020) 9743–9789. doi: 10.1021/acs.chemrev.0c00013
X. Li, S. Chen, W.D. Zhang, H.G. Hu, Chem. Rev. 120 (2020) 10079–10144. doi: 10.1021/acs.chemrev.0c00532
A.J. Cameron, P.W.R. Harris, M.A. Brimble, Angew. Chem. Int. Ed. 59 (2020) 18054–18061. doi: 10.1002/anie.202004656
Z. Wu, S. Liu, X. Cheng, et al., Chin. Chem. Lett. 27 (2016) 1731–1739. doi: 10.1016/j.cclet.2016.04.024
X. Li, Y. Zou, H.G. Hu, Chin. Chem. Lett. 29 (2018) 1088–1092. doi: 10.1016/j.cclet.2018.01.018
K. Rosenthal-Aizman, G. Svensson, A. Unden, J. Am. Chem. Soc. 126 (2004) 3372–3373. doi: 10.1021/ja0372659
B. Li, X. Li, B. Han, et al., J. Am. Chem. Soc. 141 (2019) 9401–9407. doi: 10.1021/jacs.9b04221
A.D. de Araujo, H.N. Hoang, W.M. Kok, et al., Angew. Chem. Int. Ed. 53 (2014) 6965–6969. doi: 10.1002/anie.201310245
X.X. Chen, Y. Tang, M. Wu, et al., Org. Lett. 23 (2021) 7792–7796. doi: 10.1021/acs.orglett.1c02820

Figure 2 Scope of on-resin peptide modification (a) and fragment condensation (b). Reaction conditions: resin-bonded 1 (0.2 mmol from resin loading), 2 (0.4 mmol), DMF (10.0 mL), 85 ℃, 24 h, isolated yield calculated from resin loading. a In-solution fragment condensation: fully protected peptide 1 (0.2 mmol), 2 (0.2 mmol), MeCN (10.0 mL), 85 ℃, 24 h, isolated yield.

Figure 3 On-resin cyclization. Reaction conditions: 2-Bromoacetylated 4 (0.2 mmol from resin loading), DMF (25.0 mL), 85 ℃, 24 h, total yield from resin loading. a Stapling precursor was prepared from acetylated 4 (0.2 mmol from resin loading), isolated yield calculated from resin loading.

Figure 4 (A) Chemical structures of 6 and 7. (B) Fluorescent confocal microscopy images of the HeLa cells treated with 10 µmol/L FITC labeled peptides for 12 h.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载: