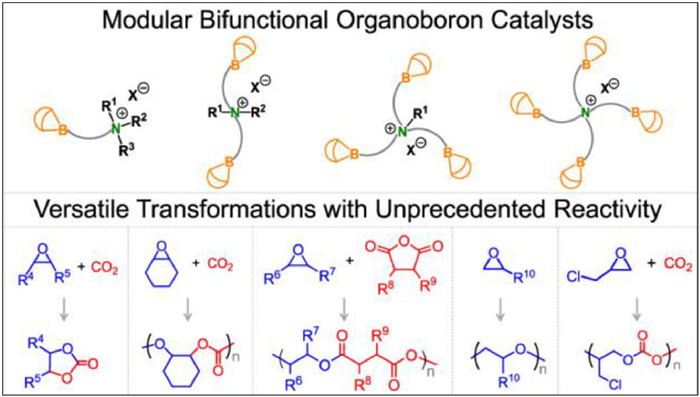
Figure 1.
Versatile organoboron catalysts for ring-opening (co)polymerizations. Reproduced with permission [2]. Copyright 2021, American Chemical Society.
Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2021
Quan Wen , Qiuquan Cai , Ping Fu , Dan Chang , Xiaoyi Xu , Tian-Jiao Wen , Guang-Peng Wu , Weipu Zhu , Ling-Shu Wan , Chengjian Zhang , Xing-Hong Zhang , Qiao Jin , Zi-Liang Wu , Chao Gao , Haoke Zhang , Ning Huang , Chang-Zhi Li , Hanying Li

The MOE Key Laboratory of Macromolecular Synthesis and Functionalization was established in Zhejiang University, focusing on the fundamental and applied polymer science, including controllable catalytic polymerization, microstructure and rheology, photo-electro-magnetic functional polymers, biomedical functional polymers and separating functional polymers [1].
In 2021, the laboratory has achieved outstanding results in the five main research directions mentioned above, which are worth highlighting. Here, we wish to present eleven published studies in this review, including versatile organoboron catalysts, catalyst-free polyesters, engineered surface coatings, polymer synthesis, drug delivery, shape-memory glassy hydrogels, reversible fusion and fission of wet-spun graphene oxide fibers, clusteroluminescent polypeptides, conductive covalent organic frameworks, polymer solar cells and long-range ordered bulk-heterojunctions.
The past two decades have witnessed tremendous progress in organocatalysts and their widespread use in producing fine chemicals and polymeric materials, because of their immense potential of savings in cost, time, and energy, as well as reductions in chemical waste. More recently, our group reported a series of mono-, di-, tri-, and tetranuclear organoboron catalysts with unprecedented activities for several chemical transformations [1,2]. Last year, we highlighted this kind of catalysts for copolymerization of CO2/cyclohexane oxide (CHO) [3], cycloaddition of CO2 and epoxides [4], and ring-opening polymerization (ROP) of propylene oxide and ethylene oxide [5], herein, in this article, we continue our efforts and share this versatile catalysts for ROPs and ring-opening copolymerizations (ROCOP) to produce polycarbonate and polyesters (Fig. 1).
Ring-opening polymerization affords an efficient strategy to convert β-butyrolactone into polyhydroxyalkanoates (PHAs) which is a promising and widely used biodegradable polyester. To expand the feasibility of our catalysts and provide a metal-free catalytic system to deliver poly(β-hydroxybutyrate) (PHB) in a controlled manner, last year, we made our efforts to optimize the organoboron catalyst with good reactivity, excellent polymer selectivity, and a controlled property for polymerizing β-butyrolactone [6]. In addition, the carbonylative polymerization of epoxides provides a promising but challenging strategy to synthesize PHAs which are of high commercial value in the field of biomedical materials and engineering plastics. Last year, we further developed a series of well-defined bifunctional organoboron-cobalt catalysts, combined advantages of simple preparation, high yields, and low metal content for the direct preparation of PHA oligomers from epoxides and CO. The organoboron-cobalt-mediated carbonylative polymerization exhibits high polyester selectivity (over 95%, four epoxide examples), and the obtained PHAs have more than 99% ester linkages on the polymer main chain [7].
ROCOP of epoxides with cyclic anhydrides is a useful method to produce polyesters, but the product with high molecular weight (Mn) remains a major challenge. In 2021, we communicated a metal-free, highly active, and high thermoresistance system for the ROCOP of epoxides with cyclic anhydrides to prepare polyesters (13 examples) [5]. The organoboron catalysts can endure a reaction temperature as high as 180 ℃ for the ROCOP of CHO with phthalic anhydride (PA) without the observation of any side reactions. The average Mn of the produced poly(CHO-alt-PA) climbed to 94.5 kDa with low polydispersity (Ð = 1.19). Furthermore, we achieved an unprecedented efficiency of 7.4 kg of polyester/g of catalyst, at a feed ratio of CHO/PA/catalyst = 20000:10000:1 at 150 ℃ [5].
The exploration of copolymerization of epichlorohydrin (ECH)/CO2 to produce poly(chloropropylene carbonate) (PCPC) is a challenging topic, because of the small energy difference arises from the electron-withdrawing chloromethyl group, which leads to a strong tendency for cyclic carbonate byproduct formation rather than the desired polycarbonate. Last year, we provided kinds of pinwheel-shaped tetranuclear organoboron catalysts for ECH/CO2 copolymerization with > 99% polymer selectivity and quantitative CO2 uptake (> 99% carbonate linkages) under mild conditions (25-40 ℃, 25 bar of CO2) [8].
In short, the metal-free feature, high catalytic performance under mild conditions, and no trouble with chromaticity for the produced polymers imply that our catalysts are practical candidates to advance polymer science.
As an essential living subsistence, polyester has covered the fields of fabric fibers, catering utensils, medical instruments, and so forth, with a global annual consumption of over 70 million tons [9]. Currently, melt polycondensation using low-cost commercial dicarboxylic acid and diol monomers is the most economical way for large-scale production of polyesters [1,10-12]. Metal catalysts are commonly employed in the polyester production to accelerate the reaction rate and increase the molecular weight of the product [13]. However, the highly biotoxic metal ions of these catalysts, e.g., Sb, Sn, Ge and Ni, will be left in the polyester matrix, posing a serious threat to the human body and natural environment [14,15]. As early as 1929, Wallace H. Carothers, the pioneer of polymer science, had reported that dicarboxylic acid and diol could undergo an esterification autocatalyzed by the diacid monomer to afford the polyester without any catalyst residue. Unfortunately, the molecular weight of the product was only 2-5 kDa, making it unpractical due to its poor performance [16,17]. Yet this strategy to produce catalyst-free polyesters is still of great interest nearly a century later.
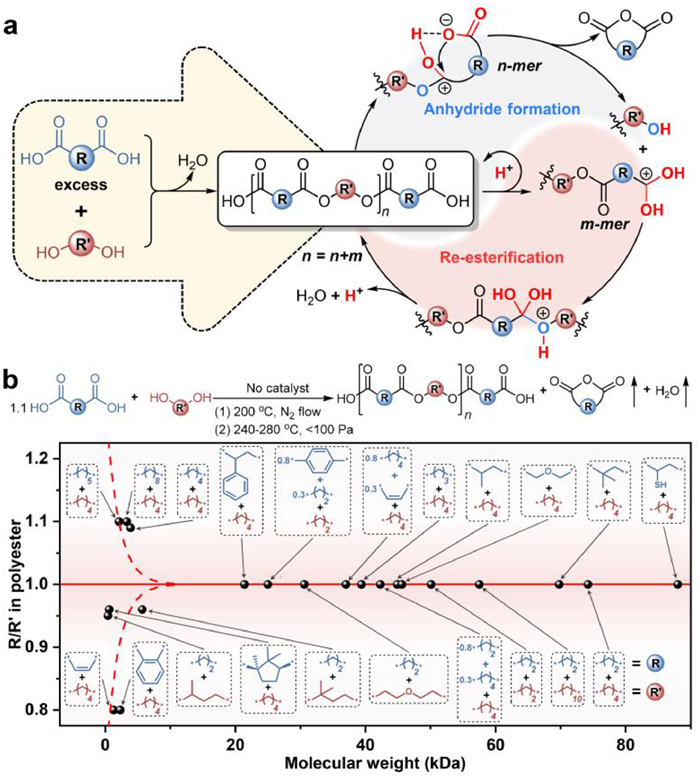
The challenge lies in how to figure out the real reason why it is difficult to obtain high molecular weight (HMW) polyester by autocatalysis. Thermodynamic factors, i.e., the low equilibrium constant of esterification and the difficulty in excluding water as a by-product at high melt viscosity, have been widely considered to be the reason [10]. Nevertheless, a transesterification strategy widely applied in industry can obtain HMW polyesters using metal catalysts, where the equilibrium constant (< 1) of transesterification is even smaller than that of esterification (~4); the by-product, i.e., ethylene glycol (EG), is more difficult to be removed than water [18-20]. These results indicate that thermodynamic factors should not be the real reason for the low molecular weight of the product under autocatalysis. Actually, the core problem addressed by transesterification is to prevent the kinetic deviation of the diacid/diol monomers from the equimolar ratio, a key factor that is often overlooked leading to low molecular weight products [21-27]. Hence, the kinetic factor should be the key to limit the molecular weight growth of polyesters via autocatalysis.
Accordingly, we proposed a catalyst-free polycondensation (CFP) mechanism using a series of dicarboxylic acids capable of forming five- or six-membered cyclic anhydrides as monomers (Fig. 2a). When excess diacid is esterified with primary diol to yield a carboxyl-terminated prepolymer, the following tandem reactions will occur: (1) The carboxyl-terminated prepolymer undergoes reversible intramolecular and intermolecular proton transfer to form carboxyl cations and carboxyl zwitterions, respectively; (2) The carboxyl zwitterions generate cyclic anhydrides through chain-end back-biting to release reactive hydroxyl groups; (3) The regenerated hydroxyl groups and carboxyl cations are re-esterified to propagate the polymer chain. Thus, the diacid/diol composition ratio (R/R') of polyesters can constantly approach 1:1, thereby enabling HMW catalyst-free polyesters (Fig. 2b). We confirmed that the reaction of various anhydride-formable diacids, e.g., (±)-phenylsuccinic acid, glutaric acid, (±)-methylsuccinic acid, diglycolic acid, 2,2-dimethylsuccinic acid, succinic acid (SA), and (±)-mercaptosuccinic acid, with 1,4-butanediol (BDO), as well as the reaction of SA with a variety of primary diols, e.g., diethylene glycol, EG, and 1,10-decanediol, can produce polyesters with molecular weights as high as 21.4-88.0 kDa. By contrast, diacids with long-chain structures such as adipic acid (AA), pimelic acid, and sebacic acid, will always have an R/R' value greater than 1:1 due to the extremely slow rate of anhydride formation relative to re-esterification, thus failing to obtain HMW products. However, the excessively fast anhydride formation compared to re-esterification also results in R/R' being less than 1:1, still leading to oligomers. These cases include the use of sterically hindered diacids such as maleic acid (MA), phthalic acid, and (+)-camphoric acid to react with BDO, or SA to react with non-primary diols, e.g., 1,3-butanediol and (±)-3-methyl-1,3-butanediol. Furthermore, the CFP can be extended to diacids that are difficult or impossible to form anhydrides by copolycondensation. For example, SA/AA/BDO, AA/MA/BDO, terephthalic acid/SA/EG can obtain HMW copolyesters with the feed ratio of 0.8:0.3:1 [28]. Through ingeniously designed comonomers, the CFP can also be employed to synthesize copolyesters with antibacterial properties [29].
Surface treatment plays an important role in materials science and engineering. Various methods such as plasma treatment [30], self-assembled monolayer [31], and layer-by-layer self-assembly [32] have been intensively studied. In 2007, mussel-inspired surface chemistry based on polydopamine was developed for multifunctional coatings [33]. This simple strategy enables surface modification under mild conditions for a wide variety of substrates including both organic and inorganic materials, and it also provides a versatile platform for post-functionalization due to the abundant functional groups on the coatings [34]. The deposition process can be accelerated and the coating uniformity can be improved by a co-deposition strategy with amino-rich compounds such as polyethyleneimine [35]. Based on the dopamine chemistry, we have already made some progress in Janus polymer membrane preparation [36] and isoporous membrane surface modification [37]. On the other hand, plant polyphenols that found in tea, chocolate, and wine, represented by tannic acid, can form colorless multifunctional coatings [38]. These polyphenols can deposit on various substrates rapidly through one-step coordination with metal ions to form metal-phenolic networks [39].
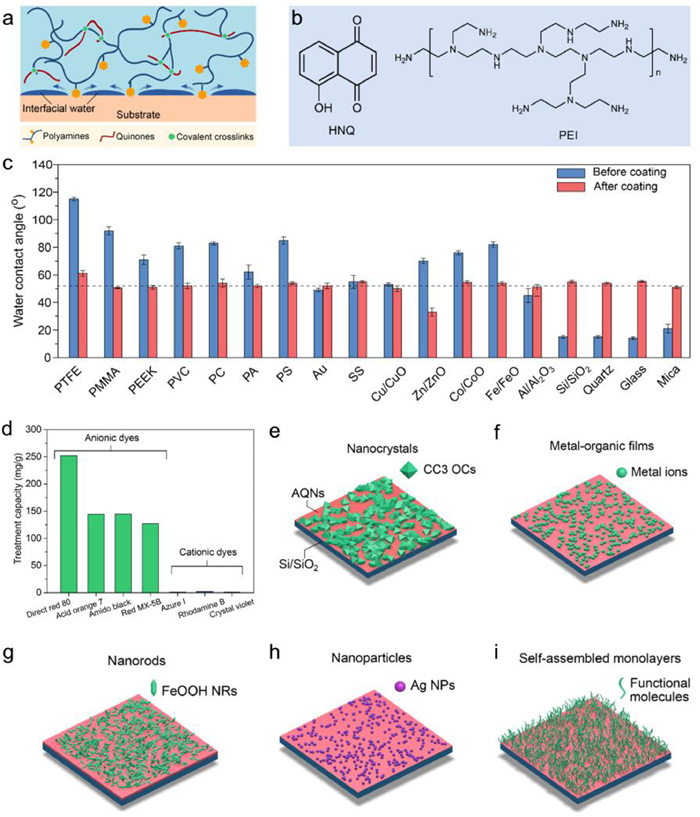
It is widely accepted that the catechol structure in polyphenols is essential for the effective formation of surface coatings. However, similar coatings, inspired by juglone that has no catechol structure, can also be created through the assembly of metal-quinone networks or amino-quinone networks. We have reported the coordination of 5-hydroxy-1,4-naphthoquinone (HNQ), also called juglone, with FeⅢ to form HNQ-FeⅢ networks on various substrates [40]. While by the assembly of HNQ with polyamines, we also prepared robust and substrate-independent surface coatings with tunable structure and physicochemical properties such as thickness, adhesion, wettability, and surface charge (Figs. 3a and b) [41]. We proposed that the positively charged amino groups in polyamines can remove the hydration layer of the substrate rapidly, and the quinone displays non-covalent interactions such as π-π stacking on polymer surface and metal coordination on metal oxide surface. The amine building blocks play a vital role in the deposition process: with increased amino groups in the amine molecules, the adhesion force is greatly changed and the resultant film thickness and optical transmittance can be tailored. Besides, due to the hydrophilicity and positive charge of amine groups, we can modulate both surface wettability and surface charges of the modified substrate (Fig. 3c). Microporous polypropylene membranes functionalized with amino-quinone networks can selectively adsorb anionic dyes from water by taking advantage of the positively charged property of the modified surface (Fig. 3d). In addition, owing to the multiple functional groups, the coatings can mediate many secondary reactions such as liquid-phase epitaxy of crystals (e.g., crystalline (1S, 2S)-1,2-cyclohexane diamine cage, CC3), preparation of metal-organic films by chelating metal ions (e.g., FeⅢ, CuⅡ and ZnⅡ), controlled growth of various nanomaterials (e.g., FeOOH nanorods and Ag nanoparticles), and self-assembly of monolayers of functional molecules (e.g., thiol-containing molecules and catechol-containing molecules) (Figs. 3e-i).
Phenols without catechol structures as building blocks provide a platform to construct more extensive phenolic networks. We also found some other novel surface coatings based on vanillic and ferulic acid, both of which have just one phenolic hydroxyl group [42-44]. They can coordinate with metal ions such as FeⅢ and CuⅡ to form metal-phenolic networks and achieve rapid surface modification for both organic and inorganic substrates. We believe these findings greatly expand the building blocks for novel surface coatings.
Plastic pollution has become one of the most pressing environmental concerns, with the rapid growth of single-use non-degradable plastic products [45-47]. The pressure of plastic pollution has prompted researchers to look for biodegradable polymers to replace polyolefin materials in certain applications [48,49]. Aliphatic polycarbonate and polyester are the most important two types of biodegradable materials, which are also biocompatible and show great application prospects in the field of biomedicine [50-52]. However, their practical application is greatly limited in terms of high cost and poor performance. Developing simple and efficient synthetic catalysts and improving the properties of polymers are the two main research directions in the field of polycarbonate and polyester.
The copolymerization of CO2 with propylene oxide (PO) can obtain poly(propylene carbonate) (PPC) that has good mechanical properties and excellent oxygen barrier properties [53]. The core challenge is to develop low-cost and highly effective catalysts for CO2-PO copolymerization. In the past decade, a variety of high-efficient metal catalysts bearing subtle ligands have been developed for the copolymerization [54-58]. However, complex synthesis procedures for the catalysts are typically required. Furthermore, the metal and ligand residues in the product are difficult to remove [59,60]. In 2016, Gnanou, Feng, and co-workers reported the first example of organocatalytic CO2-PO copolymerization using triethyl borane (TEB) combined with onium halides or alkoxides, affording metal-free PPCs with alternating degrees (ADs) of 73%-99% with PO conversion of 19%-87% at 60 ℃ within 10 h [61]. We disclosed the B-N Lewis pair composed of small molecules of TEB and triethylamine (TEA) for CO2-PO copolymerization (Fig. 4a), exhibiting high productivity of 171 g PPC/g catalyst [62]. A diamine, N,N,N′,N′-tetraethyl ethylenediamine combined with TEB exhibited productivity up to 216 g PPC/g catalyst. Obtained PPCs have AD up to 99%, selectivity (vs. propylene carbonate) up to 99%, head-to-tail linkages of 80%-82%, Mn up to 56.0 kDa, and Ð < 1.2. For the mechanism, successive insertion of PO and CO2 into the B-N Lewis pair leads to the formation of an end-to-end zwitterion featuring TEB-masked anion and onium cation. The overall catalytic performance of the B-N pair is better than other reported organocatalysts for CO2-PO copolymerization.
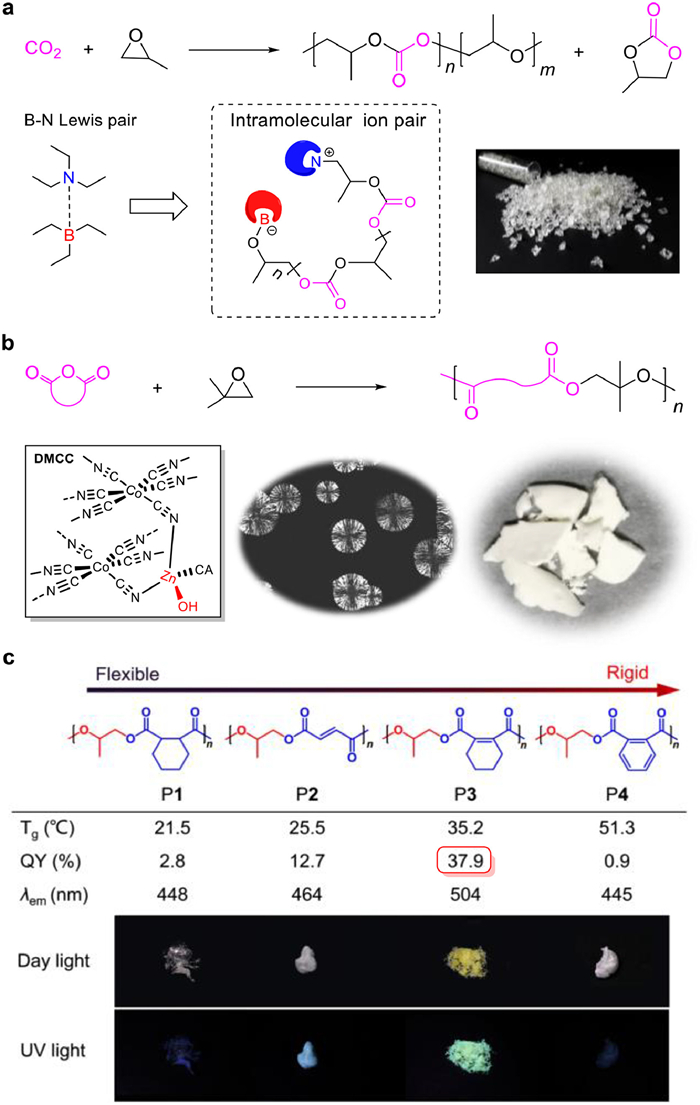
Alternating copolymerization of epoxides with cyclic anhydrides (CO2 is the carbonic anhydride with only one carbon) is an attractive chain-growth route to produce polyesters [63]. To date, more than 400 polyesters have been discovered by this method. However, the semicrystalline polyester obtained from this route is still rare [64]. Crystallization can enable polymers with enhanced physical and mechanical properties [65]. We described a facile approach to semicrystalline polyesters via the copolymerization of isobutylene oxide (IBO) and cyclic anhydrides (Fig. 4b) [66]. The heterogeneous catalyst of zinc-cobalt(Ⅲ) double metal cyanide complex (DMCC) can effectively inhibit IBO isomerization and exhibits high productivity up to 680 g polyester/g catalyst. Obtained polyesters have AD of > 99%, head-to-tail linkages of > 90%, and Mn up to 42.3 kDa. This method also produced low-molecular-weight (3.9-7.1 kDa) telechelic polyesters with hydroxyl terminals by adding water as a chain transfer agent. Obtained polyesters are semicrystalline owing to high regioregularity and exhibit melting temperatures of 67-141 ℃. Especially, the polyester prepared from IBO and maleic anhydride (MA) exhibits a crystalline-amorphous-crystalline transformation by tuning cis/trans isomerization of C=C bonds.
Organic fluorescent materials have been widely investigated for many advanced applications such as sensors, cell imaging, electroluminescence, and display technology [67-71]. Based on the DMCC catalysis for the synthesis of polyesters, we developed the first example of linear non-conjugated polyester showing yellow-green clusteroluminescence (CL, Fig. 4c) [72]. We synthesized four polyesters (P1-P4) via copolymerization of PO with cyclic anhydrides. Of special, the polyester P3 with balanced flexibility and rigidity showed the longest CL wavelength (504 nm) and highest quantum yield (QY, 38%). The systematically photophysical characterization of the system revealed that the clusters formed through the aggregate of ester units. We also demonstrated P3 as a highly selective and sensitive detector (detection limit is 0.78 mmol/L) for irons owing to the fast disassociation of clusters by irons. This work provides a new strategy to design high-efficiency and long wavelength CL and enlightens the potential application of luminescent polyesters.
Supramolecular drug delivery systems (SDDSs), which are fabricated by noncovalent interactions have received considerable attention owing to the dynamically switchable structure, special physical/chemical properties, and controlled assembly/disassembly [73-78]. By manipulating the assembly/disassembly process, smart drug release could be achieved in response to various stimuli, such as pH, enzymes, light, temperature, which provides a versatile nanoplatform as functional drug nanocarriers [79-84].
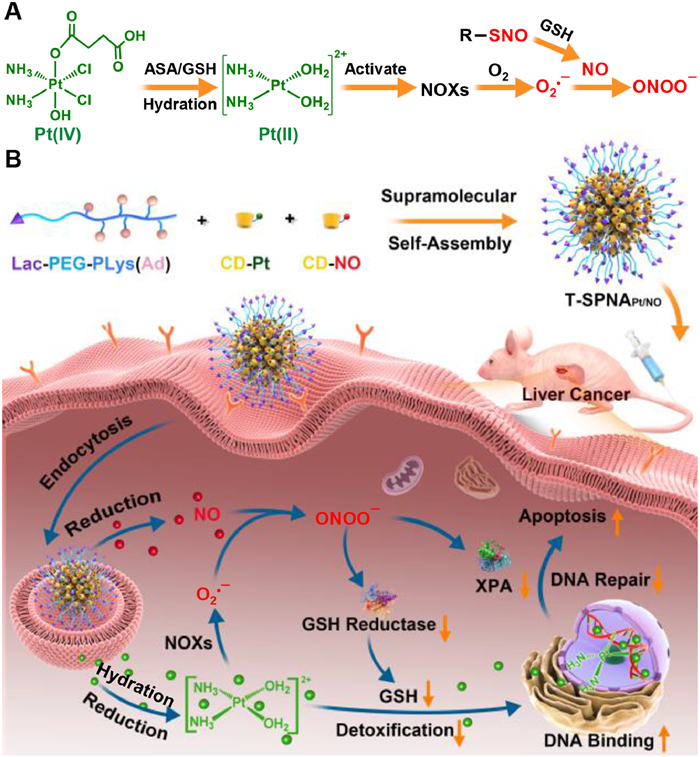
Cyclodextrin (CD) is an important complexing agent in the pharmaceutical industry, which is mainly used to increase the aqueous solubility and improve the stability of drugs [85]. Moreover, CD is the most widely used host molecule in fabricating SDDSs, which can be utilized to form inclusion complexes with many guest molecules by host-guest interactions, including polyethylene glycol (PEG), adamantane (Ad), ferrocene, azobenzene, and cholesterol [86,87]. Taking advantage of the host-guest interaction between β-CD and Ad, we fabricated a supramolecular pro-drug nanoassembly (SPNA, Fig. 5) for the co-delivery of cisplatin (CDDP) and nitric oxide (NO) [88]. We prepared the lactose targeted SPNA (T-SPNA) by the self-asembly of lactose-ended poly(ethylene glycol)-block-polylysine with pendant adamantane (Lac-PEG-b-PLys(Ad)), and two β-CD based prodrugs (Pt(Ⅳ) conjugated β-CD (CD-Pt) and NO donor conjugated β-CD (CD-NO)). It is interesting to find that we can adjust the composition of Pt and NO in SPNAPt/NO precisely by controlling CD-Pt and CD-NO in feed, which implies that such SDDS is very advantageous to fabricate drug co-delivery systems compared to conventional drug delivery systems. After internalized by liver cancer cells, both Pt and NO could be released from T-SPNAPt/NO in intracellular reductive environment. Pt was known to activate nicotinamide adenine dinucleotide phosphate oxidases (NOXs) to produce O2•−, which could further react with NO to generate peroxynitrite anion (ONOO−). ONOO− shows stronger oxidation and nitration capability than most reactive oxygen species (ROS) and reactive nitrogen species (RNS). Because of the production of ONOO−, the intracellular glutathione reductase (GR) level was greatly down-regulated, leading to the decrease of intracellular glutathione (GSH) level. As is known, GSH can form Pt-GSH adducts with Pt and induce the detoxification of Pt(Ⅱ), which can promote the efflux of intracellular Pt(Ⅱ) and reduce intracellular Pt-DNA adducts. The decrease of intracellular GSH level by ONOO− significantly increase the cytotoxicity and anti-tumor capability of CDDP. At the same time, ONOO− can also reduce the expression of an essential DNA damage repair protein, xeroderma pigmentosum group A (XPA), to block the repair of damaged DNA. It is known that the DNA damage repair is the main mechanism of CDDP-induced resistance. Therefore, the generation of ONOO− by the integration of NO into the CDDP delivery system could effectively overcome CDDP-induced drug resistance. We confirmed the ONOO− potentiated chemotherapy of CDDP on different liver cancer models, including a subcutaneous LM3 hepatoma model, an orthotopic CDDP-resistance hepatoma model, and a subcutaneous patient-derived hepatoma xenograft model. This research offers an innovative supramolecular co-delivery strategy for RNS-potentiated chemotherapy, which is very promising for RNS-mediated cancer therapy.
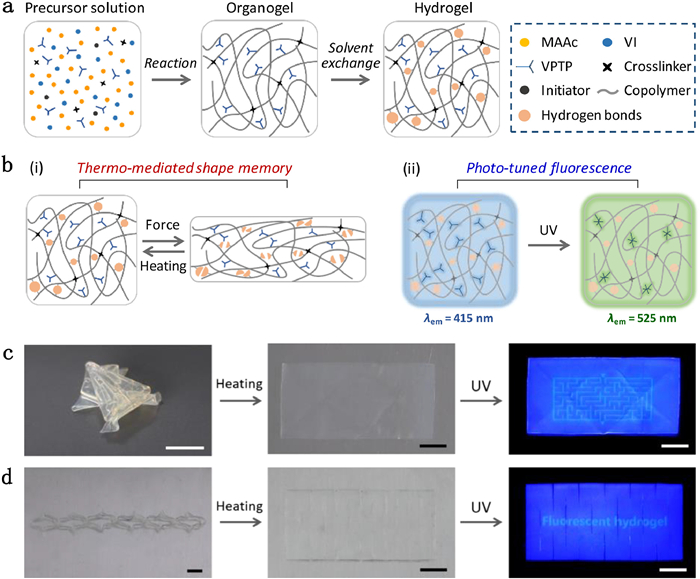
Hydrogels have received tremendous interest due to their close similarities to soft biotissues, multiple responses to external stimuli, and versatile applications in tissue engineering, drug delivery, flexible electronics, soft actuators/robots, etc. [89-94]. However, conventional hydrogels are usually mechanically weak, limiting their applications in load-bearing conditions. In the past two decades, many efforts are devoted to improving the mechanical properties of hydrogels. Various tough hydrogels have been developed by controlling the network structures and imparting energy dissipation mechanisms [95-100]. Some hydrogels have tensile breaking stress of ~10 MPa and fracture energy of ~9000 J/m2, comparable to those of biological tissues [95,96]. However, in terms of stiffness, the synthetic tough hydrogels with Young's modulus of 0.1-1 MPa are much softer than some biotissues such as cartilages and ligaments (modulus of 10-100 MPa). These soft hydrogels will experience considerable deformations under mechanical loading, limiting the applications as structural elements. It is challenging yet highly desired to develop stiff and tough hydrogels with large amount of water.
Recently, we prepared a series of tough and stiff supramolecular hydrogels based on the formation of dense and robust associative interactions [101,102]. For example, The poly(methacrylamide-co-methacrylic acid) hydrogels with water content of 50-70 wt% had Young's modulus of 10-100 MPa, tensile breaking stress of 1-10 MPa, and fracture energy of 5-20 kJ/m2 [101]. Typical forced elastic deformation was observed, accompanying with yielding, stress whitening, and crazing phenomena. Dynamic mechanical analysis revealed that these gels were in the glassy state at room temperature. The reduced mobility of chain segments and dynamics of the networks were related to the robust hydrogen bonds between the carboxylic acid and acrylamide groups, which were stabilized and enhanced by the hydrophobic α-methyl groups. The contributions of chain rigidity, entanglements, and hydrogen-bond associations were examined by rheological measurements and tensile tests of the poly(acrylamide-co-methacrylic acid) hydrogels with different compositions [103]. The synergic effect of hydrogen bonding and hydrophobic interactions led to associations with wide distribution of strength and therefore a broad viscoelastic transition in the rheological dynamic spectra of the tough gels. The design principle has been applied to various systems to prepare glassy hydrogels by forming dense and robust associative interactions [104-106], providing a facile avenue to develop hydrogels with balanced mechanical properties.
Owing to the dynamic nature of these associative interactions, the mechanical properties of these transparent glassy hydrogels depended on test temperature [101,102,104]. The modulus drastically decreased as the temperature across the glass transition of the hydrogel. The reversible temperature-mediated variations of mechanical properties, especially the Young's modulus, afforded the hydrogels with good shape memory properties. We applied this shape-memory hydrogel for dual encryption of fluorescent information [107]. As shown in Fig. 6, we fabricated the fluorescent shape-memory hydrogel by incorporating chromophore unit of 4′-(N-vinyl benzyl-4-pyridinyl)-2,2′:6′,2′′-terpyridine perchlorate (VPTP) into poly(1-vinylimidazole-co-methacrylic acid) (P(VI-co-MAAc)) matrix. The dense hydrogen bonds between the imidazole and acrylic acid groups rendered the hydrogel with excellent mechanical properties and temperature-mediated shape-memory effect. Meanwhile, this hydrogel also showed tunable fluorescence because of the photo-induced transformation of the chromophore units from unimer to dimer form, resulting in shift of the fluorescent color from blue to green. Fluorescent information was encoded by photolithography to locally tune the chromophore state and the fluorescent color. Then, this hydrogel was deformed into specific three-dimensional configurations to complete the information encryption by harnessing the shape-memory effect. The concealed information in the architected hydrogels became readable only after the sequential shape recovery and UV light irradiation. We believe that these glassy hydrogels with excellent mechanical performances and shape-memory properties should find other applications in artificial organs, biomedical devices, soft actuators, etc. [108,109].
The discovery of novel interfacial assembly property of atoms and molecules is the key to unlocking their applications. According to previous reports, multiple artificial assemblies of metal atoms or amphiphilic small molecules and polymer can be fused, but the fused one fails to be split in a precisely reversible manner [110-113]. That is, the number, size, chemical composition, structure, and properties of individual assemblies cannot be restored to their original state after fission. The irreversible fusion and fission behavior is caused by the interdiffusion of zero-dimensional or one-dimensional atoms/molecules at the assembled interface, which results in the unrecoverable physical or chemical changes on the structure of individual assemblies. This drawback has greatly hindered the development of intelligent materials featuring the reversible assembly ability. How to achieve the precisely reversible fusion and fission becomes a major challenge.
Aiming at the problem of reversible assembly, the team of Professor Gao Chao from Zhejiang University has been searching for suitable solutions. This research team works on the study of physicochemical properties and macroscopic assembly of graphene oxide (GO) for more than 10 years. GO is a derivative of graphene, characterized by oxygen-containing groups decorated on the skeleton of carbon rings [114-117]. On the basis of intrinsic molecular structure, GO exhibits two-dimensional topology, super-flexibility, and self-adhesive properties, which is a novel kind of two-dimensional macromolecule. In 2011, Gao's group discovered that GO sheets could spontaneously arrange in solvents to form liquid crystal phase as the concentration exceeded a certain value [118]. The discovery and control of GO liquid crystal facilitate the assembly of GO into macroscopic materials, for example, GO fibers [119-121]. In 2016, Gao's team firstly reported the self-fusion effect of GO fibers [122]. When the wet-spun GO gel fibers were deposited on the filter mesh and dried, the fibers spontaneously fused to form a multifunctional non-woven fabric. GO fibers formed X-type and Y-type connections at the nodes. However, the microscopic process and mechanism of self-fusion were unclear at that time [123].
In 2021, based on the previous research, we successfully realized the precisely reversible fusion and fission of GO fibers for the first time (Fig. 7) [124]. Under the stimulus of water or polar organic solvents, multiple macroscopic fibers fused into a thicker fiber, and the fused one split into numerous fibers that were identical to the initial single fibers (Fig. 7a). We explored the mechanism of the precise reversibility. We found that the solvent-responsive large deformation of GO fiber shell prompted self-adaptive adhesion or repulsion at fiber interface, leading to a dynamically reversible assembly interface to achieve reversible fusion and fission (Figs. 7b and c). During the fusion and fission processes, two-dimensional GO macromolecules featuring large size, high dimension and strong face-to-face interaction inhibited molecular interdiffusion at the assembled interface. These traits of GO macromolecules ensured the precise reversibility of assembly. The reversible assembly property makes novel smart applications come true. Firstly, we prepared carbon-based fibers with high mechanical strength in arbitrary diameter firstly via fusion approach. Interestingly, the mechanical strength of the fused GO fibers did not decrease with the rising diameter, which broke through the limit of Griffith's scaling law [125]. After thermal reduction, the tensile strength of fused GO fibers reached as high as 597 MPa with a large diameter of 58 µm. Secondly, we endowed GO macroscopic assemblies with the dynamic transformation ability through reversible fusion and fission. We realized intelligently reversible transformations between diverse GO fiber-based architectures, such as the transformations between a stiff rod fused by 13500 GO fibers and a flexible net with fused nodes, the transitions between a fused GO fiber and a complex GO fiber-based assembly with one-dimensional or two-dimensional structure, etc. Thirdly, we achieved controllable release of various guest compounds from GO fiber bundle. Polystyrene microspheres, sub-millimeter glass beads, and polyacrylonitrile staple fibers were included in GO fiber bundle by fusion, and controllably expelled from the bundle by fission. Finally, we readily extended the reversible assembly capability to traditional polymeric, metallic and ceramic fibers, by constructing a coating of GO on their surface. This advantage further expanded the application range of precisely reversible assembly.

This study of precisely reversible fusion and fission takes two-dimensional GO macromolecule as a model, and reveals the difference of physicochemical properties between two-dimensional molecules and other atoms and molecules. Further, we propose a universal strategy of constructing the dynamic assembly interface. This work opens up a new research direction regarding dynamic multi-functionality of artificial materials that can be strengthened, split, recombined and applicatory.
Luminescent materials, especially polymers, have many advanced applications in optoelectronic devices, environmental monitoring, explosive detection, bioimaging, etc. [126]. In the past century, mature molecular photophysical theories have been established to reveal the luminescent mechanism, which also plays an essential role in the development of new functional luminophores [127]. Thereinto, through-bond conjugation (TBC) was believed as the necessary structural and electronic factor to construct luminophores, and molecules without fused aromatic rings or extended π bonds were not supposed to fluoresce visible emission [128]. However, in recent twenty years, some aliphatic non-conjugated polymers, such as starch, cellulose, proteins and poly(amidoamine), were discovered with abnormal blue fluorescence at clustering state, namely clusteroluminescence (CL) [129]. In comparison to traditional conjugated luminophores, structures with CL (CLgens) have many advantages in practical applications, for example, good processibility and biocompatible [130]. But the thorny issue is that the TBC-based molecular photophysics is inapplicable to CLgens, which prohibited the design of new CLgens. Meanwhile, most of the reported CLgens possess short-wavelength blue emission and low fluorescence quantum yield, showing limited application potential. So, revealing the emission mechanism of CL and developing efficient CLgens not only have great theoretical significance but also show huge potential to bring a revolution to luminescent materials [131]. Driven by the above-mentioned scientific importance, we are dedicated to developing new efficient CLgens and gradually establishing new photophysical theories for CL.
Recently, we have synthesized three non-conjugated polypeptides, poly(γ-benzyl-l-glutamate) (PBLG), poly(l-glutamic acid) (PLGA) and its sodium salt (PLGA-Na) which share the same backbones but with different pendant groups [132]. As shown in Fig. 8a, these three polypeptides all show blue CL with emission maximum around 440 nm. In addition, PLGA with side chain of carboxyl shows the strongest CL intensity and fine structure of its photoluminescence (PL) spectrum, but PBLG with benzyl substitute instead of carboxyl exhibits a broad PL spectrum whose emission intensity is negligible, suggesting the important role of inter/intrachain interaction in boosting the CL intensity because of the strong hydrogen-bonding interaction in PLGA. To disclose the emitting species of 440 nm peaks in these polypeptides, we carried out systematically experimental characterization and theoretical calculation for their model compounds N-acetyl-l-alanine, N,N-diethylacetamide and ethyl acetate. These results suggested that the 440 nm CL originated from the (n,π*) transition of the mainchain amide group. Meanwhile, we also concluded that polymerization and clusterization are two effective strategies to enhance the "forbidden" (n,π*) transition. After that, summarizing the carbonyl-based aliphatic CLgens such as polyester, polyketone, polyamide, we conclude that the 440 nm emission widely exists in these systems, which should be ascribed to the (n,π*) transition of carbonyl group, and the n electron and π* orbital come from the lone-pair electrons of oxygen and C=O bond, respectively (Fig. 8b) [133]. The above results draw a clear mechanistic picture for the blue CL in carbonyl-based aliphatic polymers.
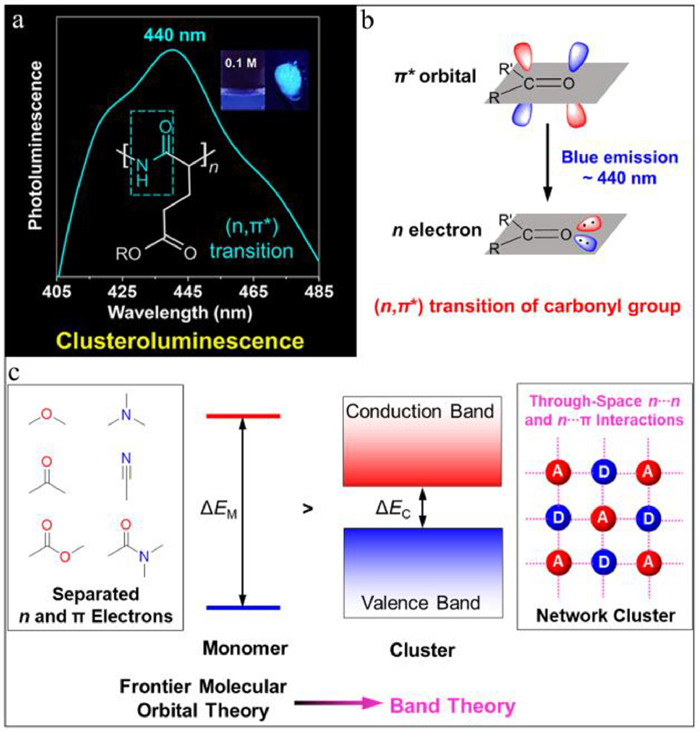
Although the emission mechanism of blue CL was revealed and the CL efficiency was successfully enhanced, it is still a big challenge to manipulate the emission color of CLgens. Then we proposed that introducing the through-space n…n, n…π and π…π interactions should be efficient strategies to adjust the bandgap of CLgens [134]. As shown in Fig. 8c, the subunits in CLgens with separated n and π electrons has a large bandgap (ΔEM) in the monodispersed state. Once forming the network clusters, their frontier molecular orbitals will be coupled and split, then the multiply splitting orbitals will form the analogous conduction and valance bands, which corresponds to a smaller bandgap (ΔEC) and bathochromic-shift CL. Based on this theory, we have manipulated the through-space π…π interaction and CL color in triphenylmethane successfully by adding electron-donating and withdrawing groups to the separated arms, especially for the donor-substituent CLgens whose maximum emission wavelength bathochromically shifted for around 45 nm, because the donors not only increased the electronic density for through-space π…π interaction but also stabilized the formed excitons [135]. In summary, we have developed new CL systems based on aliphatic polypeptides and also achieved breakthroughs in the mechanistic studies of CL. Based on the proposed theory of through-space interaction, it is promising that more and more CLgens with long-wavelength emission color and high emission efficiency will be developed and show multifunctional applications [136].
Covalent organic frameworks (COFs) have emerged as a new family of crystalline porous polymers, which are constructed by various building blocks into two-dimensional (2D) or three-dimensional (3D) periodic array structures at atomic precise [137]. In general, the 2D COFs are assembled by the stacking of layered polymeric sheets through noncovalent π-π interactions. The unique structural features endow 2D COFs with well-aligned laminar arrays and anisotropic carrier channels through intralayer and interlayer orientations [138]. In this respect, COFs are quite promising for the application in the fields of optoelectronics, sensing, electrocatalysis, and so on [139]. However, in spite of conceivable efforts, the electrical conductivity of most COFs is less than 10−8 S/cm, which is almost on the verge of insulators [140]. Therefore, the development of highly conductive COFs remains as one of the great challenges in the present or the near future.
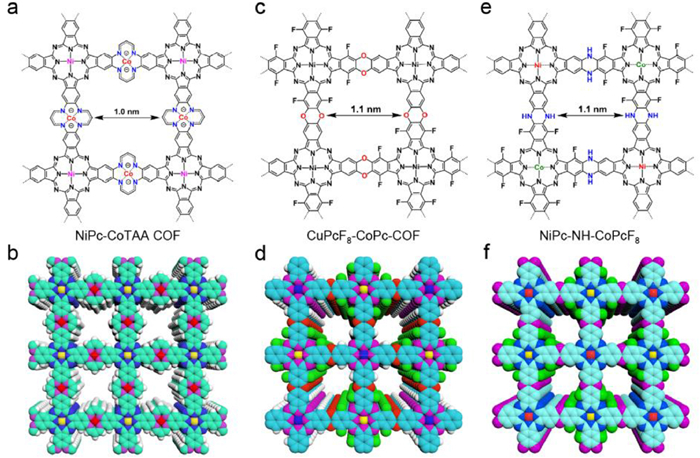
Recently, we developed a series of conductive COFs by increasing carrier density and carrier mobility. We constructed the obtained NiPc-CoTAA-COF [141], CuPcF8-CoPc-COF [142], and NiPc-NH-CoPcF8 [143] by metallophthalocyanines through tetraaza[14]annulene, dioxin, and piperazine linkages, respectively (Fig. 9). Thereinto, NiPc-CoTAA COF exhibited a highly conjugated and planar structure across the whole network combined with densely stacked metallophthalocyanine units, which could greatly facilitate the intralayer and interlayer carrier mobility (Figs. 9a and b) [141]. Notably, we can further enhance the conductivity of NiPc-CoTAA COF by doping with iodine, owing to the fact that tetraaza[14]annulene units can be oxidized to generate free radicals, thus leading to an increase in carrier density. The conductivity of iodine-doped NiPc-CoTAA reached up to 5.2 × 10−3 S/cm, which was 64 times higher than that of its pristine counterpart. Compared with NiPc-CoTAA COF, the dioxin-linked CuPcF8-CoPc-COF is non-conjugated and undoped (Figs. 9c and d) [142]. Its electrical conductivity was determined as 3.6 × 10−6 S/cm, owing to the low carrier mobility and carrier density. Significantly, the piperazine-linked NiPc-NH-CoPcF8 exhibited an electrical conductivity and a carrier mobility up to 0.13 S/cm and 35.4 cm2 V–1 s–1, which ranks as new records for conductive COFs (Figs. 9e and f) [143]. We attributed the high conductivity and carrier mobility to the neatly arrayed phthalocyanine columns and inbuilt cationic radicals. In view of the high electrical conductivity, NiPc-CoTAA COF, CuPcF8-CoPc-COF, and NiPc-NH-CoPcF8 were utilized as high-performance chemiresistor, electrocatalyst, and organic field-effect transistor, respectively. Therefore, this strategy provides new design concept to improve the electrical performance of COFs and paves the way for their application in electronic fields.
Along with the remarkable progresses being achieved in recent years [144-151], polymer solar cells (PSCs) are heading to strike the threshold as practical photovoltaic technologies, wherein the fundamental challenges remain on how to achieve the ideal figure-of-merits among the efficiency-stability-cost of PSCs. On this regard, we have recently succeeded in developing a series of non-fused electron acceptors via the concise synthesis from single aromatic units (Fig. 10a), which bear non-covalent intramolecular interaction to maintain the planarity and rigidity of molecular structure [152-156], facilitating the charge and exciton transport in condensed solid [157,158]. These fully non-fused electron acceptors show the potentially ideal combination of good photovoltaic performance and low synthetic costs of organic photovoltaic materials [159-161]. In addition, the intrinsic photostability of organic semiconductors remains hindering the durable practice of PSCs. As shown in Fig. 10b, we have further disclosed that a unique structurally volume-conserving photoisomerization of acceptor-donor-acceptor (A-D-A) acceptors serves as a surrogate towards their subsequent photochemical degradation. Along with a series of investigations, we have identified that the structural confinement to inhibit photoisomerization of these unique A-D-A non-fullerene acceptors (NFAs) from molecular level to macroscopic condensed solid which helps enhancing the photochemical stabilities of molecules, as well as the corresponding organic photovoltaic cells (OPVs) [160,162,163].

Further, we have developed high-performance eco-friendly PSCs from a feasible strategy that employs the hot spin coating of non-halogenated solutions, with power conversion efficiencies of 18.25% in o-xylene, 18.20% in p-xylene, and 18.12% in toluene, respectively [164]. The levels of performance represent the highest reported efficiency for eco-friendly organic solar cells (OSCs) at the time being report. Empowered by the newly designed quaternary blends from non-halogenated solvent processing, we have further developed high-performance, yet spectrally engineered semitransparent PSCs with power conversion efficiency (PCE) of 13.08% and a plant growth factor of 24.7% (Fig. 10c) [165]. Under the semi-transparent OSCs filtered lights, plants grow favorably and are comparable to those under fully transparent glass. It is also worthy to note that we have succeeded in fabricating large area polymer solar modules with a certified efficiency of 14.11% with respect to the aperture area (19.30 cm2) that were independently measured by Chinese National Photovoltaic Industry Metrology and Testing Center (NPVM). It has been recorded in solar efficiency table [166], representing the best organic solar minimodule so far. Overall, these combined efforts in the year of 2021 [159-165,167-173] validate that polymer solar cells possess promising perspectives as eco-friendly photovoltaics.
Improving the detection sensitivity of widely used X-ray detectors so that the detectors can operate at low-dose rates is important for human health and high-quality imaging [174]. There are mainly two improvement strategies: (1) Ordered structures of single-crystals are beneficial to charge transport, and long-range ordered lattice can ensure stable and excellent electronic properties. So high-performance X-ray direct detectors often use single-crystals as photo-responsive materials [175-177]. (2) The space charge region in a heterojunction is formed by electrons or holes diffusion in the junction area, and the separation of photogenerated electron-hole pairs is promoted by the built-in electric field [178], [179]. Therefore, we envisage combining these two strategies to construct long-range ordering bulk-heterojunctions (BHJs) to further improve the detection performance of X-ray detectors. However, traditional BHJs are composite materials with interpenetrating network heterostructures, which are difficult to achieve long-range ordering molecular arrangement [180].
We have achieved the construction of ordered BHJs by growing crystals in gel media [181-184]. Using titanate as the titanium source at room temperature, we formed a semiconducting TiO2 gel with good uniformity by sol-gel reaction. We grew PbI2 crystals by the diffusion of lead ions in the gel and iodide ions in the upper layer solution (Fig. 11a), resulting in hexagonal sheet crystals suitable for device fabrication (Fig. 11b). X-ray photoelectron spectroscopy (XPS) analysis and thermogravimetric behavior of the gel-grown crystals showed that the TiO2 gels was integrated into the PbI2 crystals (Fig. 11d) to form interpenetrating composite structure. Furthermore, the energy band extracted by ultraviolet photoelectron spectrometry (UPS) and diffuse reflectance spectroscopy (DRS) indicated that the composites are PbI2-TiO2 BHJs. Notably, powder X-ray diffraction and single-crystal X-ray diffraction revealed the long-range ordering of PbI2-TiO2 BHJs (Fig. 11c).
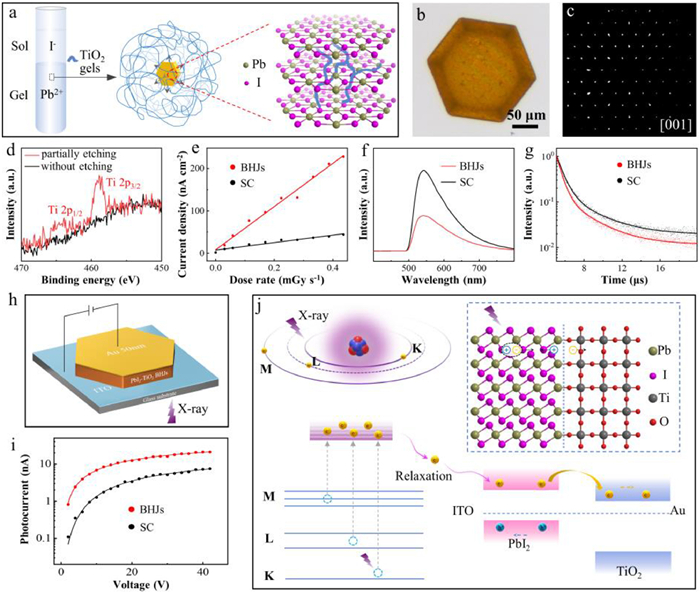
X-ray detectors were fabricated by placing BHJs single-crystals between an indium tin oxide (ITO) coated glass substrate and an Au electrode (Fig. 11h). We measured the performance as the detectors exposed to continuous X-ray irradiation with an average energy of 22 keV. Under a 15 V bias, the PbI2-TiO2 BHJs X-ray detectors exhibited a sensitivity of 509 µC Gy−1 cm−2, a 5.8 times improvement over pure single-crystals (Fig. 11e). Moreover, it is highly stable under continuous X-ray irradiation, with little change in dark- and photo-conductivity, demonstrating the structural stability of PbI2-TiO2 BHJs. The diffusion of electrons and holes in the heterojunction leads to the formation of space charge regions, triggers the transfer behavior of photogenerated charges and dissociation of electron-hole pairs. We can characterize this process by photoluminescence spectroscopy (PL). As shown in Figs. 11f and g, the reduction in PL intensity and decay lifetime reveals the enhanced carrier dissociation and transfer process, which is the key to suppress the recombination of photogenerated electron-hole pairs and improve the X-ray response charges collection. Therefore, we propose a three-step mechanism to explain the excellent X-ray detection performance of PbI2-TiO2 BHJs, as shown in Fig. 11j. The photoelectric effect mainly occurs in PbI2 crystal to generate hot electrons, including secondary electrons. They are relaxed into low-energy electron-hole pairs and consequently dissociated in the large-area space charge region. The carriers transport in the long-range ordering interpenetrating network of PbI2-TiO2 BHJs to form an X-ray response current. To confirm the mechanism, we measured the product of the mobility and lifetime (µτ) of BHJs, which is positively correlated with the diffusion distance of photogenerated charges within the semiconductor [185]. As shown in Fig. 11i, the steady-state photocurrent fitting and calculation by Hecht equation shows that the µτ of PbI2-TiO2 BHJs is 1.21 × 10−6 cm2/V, which is 6.4 times higher than that of pure single-crystal [186]. In conclusion, the combination of BHJs and long-range ordering can further improve the performance of direct X-ray detectors based on single-crystal materials.
Gel-growth of semiconductor single-crystals is simple and mild. It is a new strategy for fabricating high-performance X-ray detectors and a new platform to study the behavior of photogenerated carriers in long-range ordering BHJs. On the one hand, it is necessary to grow large-scale flat-shaped semiconductor single-crystals in gels to meet the needs of integrated devices. On the other hand, patterned gel-incorporation into semiconductor single-crystals to achieve complex device functions is an important direction of future research on single-crystals.
In conclusion, we briefly demonstrate the breakthroughs made by the laboratory in eleven research directions in 2021. The first is the design of versatile organoboron catalysts exhibiting high selectivity, high efficiency, and high thermoresistance. The second is the production of polyesters high molecular weight by catalyst-free polycondensation. The third is to achieve engineered surface coatings through surface modification, resulting in tunable structural and physicochemical properties. The fourth is the production of high-productivity of poly(propylene carbonate) and semi-crystalline polyesters and the realization of luminescent polyesters by copolymerization. The fifth is a supramolecular prodrug nanoassembly strategy to achieve RNS-potentiated chemotherapy. The sixth is a series of tough and rigid supramolecular hydrogels based on the formation of dense and robust associative interactions, with tunable fluorescence properties and good shape memory properties. The seventh is the successful first realization of reversible fusion and fission of wet-spun GO fibers. The eighth is the development of clusteroluminescent non-conjugated polypeptides and mechanistic studies. The ninth is the development of a series of conducting COFs by increasing the carrier density and carrier mobility, and the consequent high conductivity is used to enhance the performance of chemiresistors, electrocatalysts and organic field effect transistors. The tenth is the development and mechanism studies of high-performance eco-friendly polymer solar cells. The final is the mechanism of the response sensitivity improvement of long-range ordered PbI2-TiO2 BHJs X-ray detector.
For now, we are focusing on the design of more efficient and stable catalysts, optimized polymerization processes, the construction of novel surface coatings, the potential applications of luminescent polyesters, the innovative supramolecular co-delivery strategies, the application of glass hydrogels with shape memory properties, the dynamic multi-functionality of two-dimensional GO macromolecule, the multifunctional application of CLgens with high emission efficiency, the improvement of the electrical properties of COFs, the development of eco-friendly photovoltaic strategies, and the complexity of gel-crystal composites device functionalization as the focus of our future research. We believe that these functional materials and the proposed mechanism will be used to further develop new functional polymers.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors acknowledge the support from the SCI-TECH Academy of Zhejiang University.
J. Ren, X. Shu, Y. Wang, et al., Chin. Chem. Lett. 33 (2022) 1650–1658. doi: 10.1016/j.cclet.2021.10.052
G.W. Yang, Y.Y. Zhang, G.P. Wu, Acc. Chem. Res. 54 (2021) 4434–4448. doi: 10.1021/acs.accounts.1c00620
G.W. Yang, Y.Y. Zhang, R. Xie, et al., J. Am. Chem. Soc. 142 (2020) 12245–12255. doi: 10.1021/jacs.0c03651
Y.Y. Zhang, G.W. Yang, R. Xie, et al., Angew. Chem. Int. Ed. 132 (2020) 23491–23498. doi: 10.1002/ange.202010651
R. Xie, Y.Y. Zhang, G.W. Yang, et al., Angew. Chem. Int. Ed. 60 (2021) 19253–19261. doi: 10.1002/anie.202104981
L. Yang, Y.Y. Zhang, G.W. Yang, et al., Macromolecules 54 (2021) 5509–5517. doi: 10.1021/acs.macromol.1c00250
Y.Y. Zhang, L. Yang, R. Xie, et al., Macromolecules 54 (2021) 9427–9436. doi: 10.1021/acs.macromol.1c01324
G.W. Yang, C.K. Xu, R. Xie, et al., J. Am. Chem. Soc. 143 (2021) 3455–3465. doi: 10.1021/jacs.0c124250250
V. Tournier, C.M. Topham, A. Gilles, et al., Nature 580 (2020) 216–219. doi: 10.1038/s41586-020-2149-4
P.J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953.
Y.T. Yan, G. Wu, S.C. Chen, et al., Chin. Chem. Lett. 33 (2022) 2151–2154. doi: 10.1016/j.cclet.2021.08.124
Q. Ding, M. Soccio, N. Lotti, et al., Chin. J. Polym. Sci. 38 (2020) 311–322. doi: 10.1007/s10118-020-2354-5
X. Zhang, M. Fevre, G.O. Jones, et al., Chem. Rev. 118 (2018) 839–885. doi: 10.1021/acs.chemrev.7b00329
K.S. Egorova, V.P. Ananikov, Angew. Chem. Int. Ed. 55 (2016) 12150–12162. doi: 10.1002/anie.201603777
K.H. Thompson, C. Orvig, Science 300 (2003) 936–939. doi: 10.1126/science.1083004
W.H. Carothers, J. Am. Chem. Soc. 51 (1929) 2548–2559. doi: 10.1021/ja01383a041
W.H. Carothers, J. Arvin, J. Am. Chem. Soc. 51 (1929) 2560–2570. doi: 10.1021/ja01383a042
K. Tomita, H. Ida, Polymer 14 (1973) 55–60. doi: 10.1016/0032-3861(73)90096-7
T. Chen, S. Tian, Z. Xie, et al., Polym. Chem. 11 (2020) 5884–5892. doi: 10.1039/D0PY00863J
G. Challa, Recl. Trav. Chim. Pays-Bas 79 (1960) 90–100. doi: 10.1002/recl.19600790110
T. Yokozawa, A. Yokoyama, Prog. Polym. Sci. 32 (2007) 147–172. doi: 10.1016/j.progpolymsci.2006.08.001
T. Yokozawa, Y. Ohta, Chem. Rev. 116 (2016) 1950–1968. doi: 10.1021/acs.chemrev.5b00393
Q. Cai, X. Li, W. Zhu, Macromolecules 53 (2020) 2177–2186. doi: 10.1021/acs.macromol.9b02177
P. Ge, Q. Cai, H. Zhang, et al., ACS Appl. Mater. Inter. 12 (2020) 37549–37560. doi: 10.1021/acsami.0c08716
Q. Cai, J. Jiang, H. Zhang, et al., ACS Appl. Mater. Inter. 13 (2021) 19387–19397. doi: 10.1021/acsami.1c04648
Q. Cai, H. Zhang, X. Yao, et al., Acta Polym. Sin. 52 (2021) 489–498.
H. Zhang, Q. Zhang, Q. Cai, et al., Chem. Eng. J. 424 (2021) 130432. doi: 10.1016/j.cej.2021.130432
Q. Cai, T. Bai, H. Zhang, et al., Mater. Today 51 (2021) 155–164. doi: 10.1016/j.mattod.2021.07.024
H. Zhang, T. Fang, X. Yao, et al., Chem. Eng. J. 440 (2022) 135949. doi: 10.1016/j.cej.2022.135949
L.Y. Cui, Y. Yan, X.Y. Zhao, et al., Chin. Chem. Lett. 28 (2017) 1–5. doi: 10.1016/j.cclet.2016.07.028
J.C. Love, L.A. Estroff, J.K. Kriebel, et al., Chem. Rev. 105 (2005) 1103–1170. doi: 10.1021/cr0300789
C. Ye, Y. Zhu, H. Sun, et al., Chin. Chem. Lett. 32 (2021) 501–505. doi: 10.1016/j.cclet.2020.03.013
H. Lee, S.M. Dellatore, W.M. Miller, et al., Science 318 (2007) 426–430. doi: 10.1126/science.1147241
J.H. Ryu, P.B. Messersmith, H. Lee, ACS Appl, Mater. Inter. 10 (2018) 7523–7540. doi: 10.1021/acsami.7b19865
H.C. Yang, K.J. Liao, H. Huang, et al., J. Mater. Chem. A 2 (2014) 10225–10230. doi: 10.1039/C4TA00143E
G.J. Wang, B.H. Wu, Z.K. Xu, et al., Mater. Chem. Front. 3 (2019) 2102–2109. doi: 10.1039/C9QM00253G
Y. Ou, D. Zhou, Z.K. Xu, et al., Polym. Chem. 10 (2019) 3201–3209. doi: 10.1039/C9PY00560A
T.S. Sileika, D.G. Barrett, R. Zhang, et al., Angew. Chem. 125 (2013) 10966–10970. doi: 10.1002/ange.201304922
H. Ejima, J.J. Richardson, K. Liang, et al., Science 341 (2013) 154–157. doi: 10.1126/science.1237265
H. Yu, Q.Z. Zhong, T.G. Liu, et al., Langmuir 35 (2019) 3643–3650. doi: 10.1021/acs.langmuir.8b03914
Q.Z. Zhong, J.J. Richardson, A. He, et al., Angew. Chem. 133 (2021) 2376–2384.
B.P. Zhang, H.N. Li, J.L. Shen, et al., Langmuir 37 (2021) 3721–3730. doi: 10.1021/acs.langmuir.1c00221
T. An, H. Yu, L.J. Xu, et al., Acta Polym. Sin. 50 (2019) 1298–1304.
J.L. Shen, B.P. Zhang, D. Zhou, et al., Chin. Chem. Lett. 32 (2021) 3852–3856. doi: 10.1016/j.cclet.2021.05.023
J.R. Jambeck, R. Geyer, C. Wilcox, et al., Science 347 (2015) 768–771. doi: 10.1126/science.1260352
A.C. Albertsson, M. Hakkarainen, Science 358 (2017) 872–873. doi: 10.1126/science.aap8115
C. Shao, H. Duan, Y. Min, et al., Chin. Chem. Lett. 31 (2020) 299–302. doi: 10.1016/j.cclet.2019.03.053
R.A. Gross, B. Kalra, Science 297 (2002) 803–807. doi: 10.1126/science.297.5582.803
C.K. Williams, Chem. Soc. Rev. 36 (2007) 1573–1580. doi: 10.1039/b614342n
H. Tian, Z. Tang, X. Zhuang, et al., Prog. Polym. Sci. 37 (2012) 237–280. doi: 10.1016/j.progpolymsci.2011.06.004
D.J. Darensbourg, Chem. Rev. 107 (2007) 2388–2410. doi: 10.1021/cr068363q
C.J. Zhang, X. Zhang, X.H. Zhang, Sci. China Chem. 63 (2020) 1807–1814.
Y. Li, Y.Y. Zhang, B. Liu, et al., Chin. J. Polym. Sci. 36 (2018) 139–148. doi: 10.1007/s10118-018-2047-5
G.W. Coates, D.R. Moore, Angew. Chem. Int. Ed. 43 (2004) 6618–6639. doi: 10.1002/anie.200460442
X.B. Lu, D.J. Darensbourg, Chem. Soc. Rev. 41 (2012) 1462–1484. doi: 10.1039/C1CS15142H
M.R. Kember, A. Buchard, C.K. Williams, Chem. Commun. 47 (2011) 141–163. doi: 10.1039/C0CC02207A
H. Cao, Y. Qin, C. Zhuo, et al., ACS Catal. 9 (2019) 8669–8676. doi: 10.1021/acscatal.9b02741
Y. Li, Y.Y. Zhang, L.F. Hu, et al., Prog. Polym. Sci. 82 (2018) 120–157. doi: 10.1016/j.progpolymsci.2018.02.001
Y. Wang, J.L. Yang, C.J. Zhang, et al., Acta Polym. Sin. 51 (2020) 1092–1103.
C.J. Zhang, X.H. Zhang, Sci. China Chem. 62 (2019) 1087–1089.
D. Zhang, S.K. Boopathi, N. Hadjichristidis, et al., J. Am. Chem. Soc. 138 (2016) 11117–11120. doi: 10.1021/jacs.6b06679
Y. Wang, J.Y. Zhang, J.L. Yang, et al., Macromolecules 54 (2021) 2178–2186. doi: 10.1021/acs.macromol.0c02377
J.M. Longo, M.J. Sanford, G.W. Coates, Chem. Rev. 116 (2016) 15167–15197. doi: 10.1021/acs.chemrev.6b00553
Z.Q. Wan, J.M. Longo, L.X. Liang, et al., J. Am. Chem. Soc. 141 (2019) 14780–14787. doi: 10.1021/jacs.9b07058
B. Lotz, T. Miyoshi, S.Z. Cheng, Macromolecules 50 (2017) 5995–6025. doi: 10.1021/acs.macromol.7b00907
L. Hu, X. Zhang, X. Cao, et al., Macromolecules 54 (2021) 6182–6190. doi: 10.1021/acs.macromol.1c00793
W. Cao, F. Dai, R. Hu, et al., J. Am. Chem. Soc. 142 (2019) 978–986.
S.H. Hwang, Adv. Mater. 27 (2015) 2515–2520. doi: 10.1002/adma.201500267
S. Jiang, L. Meng, W. Ma, et al., Chin. Chem. Lett. 32 (2021) 1037–1040. doi: 10.1016/j.cclet.2020.09.019
S. Chen, L. Yin, L. Liu, et al., Chin. Chem. Lett. 32 (2021) 3133–3136. doi: 10.1016/j.cclet.2021.03.059
Y. Tang, S. Cabrini, J. Nie, et al., Chin. Chem. Lett. 31 (2020) 256–260. doi: 10.1016/j.cclet.2019.04.012
B. Chu, H. Zhang, L. Hu, et al., Angew. Chem. Int. Ed. 61 (2022) e202114117. doi: 10.1002/anie.202114117
S. Li, Y. Gao, Y. Ding, et al., Chin. Chem. Lett. 32 (2021) 313–318. doi: 10.1016/j.cclet.2020.04.049
M.J. Webber, R. Langer, Chem. Soc. Rev. 46 (2017) 6600–6620. doi: 10.1039/C7CS00391A
Y. Deng, P. Song, X. Chen, et al., ACS Nano 14 (2020) 9711–9727. doi: 10.1021/acsnano.0c01350
X. Chen, H. Gao, Y. Deng, et al., ACS Nano 14 (2020) 5121–5134. doi: 10.1021/acsnano.0c02197
Y. Gao, Y. Gao, Y. Ding, et al., Chin. Chem. Lett. 32 (2021) 949–953. doi: 10.1016/j.cclet.2020.08.010
Y. Liu, S. Jiang, W. Mao, et al., Chin. Chem. Lett. 33 (2022) 209–212. doi: 10.1016/j.cclet.2021.07.024
Y. Cai, Z. Zhang, Y. Ding, et al., Chin. Chem. Lett. 32 (2021) 1267–1279. doi: 10.1016/j.cclet.2020.10.036
W. Zhang, X.F. Du, B. Liu, et al., ACS Nano 16 (2021) 1421–1435.
C. Liu, Y. Xia, Z. Tao, et al., Chin. Chem. Lett. 33 (2022) 1529–1532. doi: 10.1016/j.cclet.2021.08.108
Y. Zhang, L. Wang, J. Wang, et al., Chin. Chem. Lett. 32 (2021) 1902–1906. doi: 10.1016/j.cclet.2021.01.032
Z. Li, N. Song, Y.W. Yang, Matter 1 (2019) 345–368. doi: 10.1016/j.matt.2019.05.019
P. Xing, Y. Zhao, Small Methods 2 (2018) 1700364. doi: 10.1002/smtd.201700364
T. Loftsson, D. Duchene, Int. J. Pharm. 329 (2007) 1–11. doi: 10.1016/j.ijpharm.2006.10.044
J. Zhang, P.X. Ma, Adv. Drug Deliver. Rev. 65 (2013) 1215–1233. doi: 10.1016/j.addr.2013.05.001
D. Jia, X. Ma, Y. Lu, et al., Chin. Chem. Lett. 32 (2021) 162–167. doi: 10.1016/j.cclet.2020.11.052
Y. Deng, Y. Wang, F. Jia, et al., ACS Nano 15 (2021) 8663–8675. doi: 10.1021/acsnano.1c00698
X. Liu, J. Liu, S. Lin, et al., Mater. Today 36 (2020) 102–124. doi: 10.1016/j.mattod.2019.12.026
A. Sun, X. He, X. Ji, et al., Chin. Chem. Lett. 32 (2021) 2117–2126. doi: 10.1016/j.cclet.2021.01.048
A. Zhang, F. Wang, L. Chen, et al., Chin. Chem. Lett. 32 (2021) 2923–2932. doi: 10.1016/j.cclet.2021.03.073
M. Wu, Q.Y. Peng, L.B. Han, et al., Chin. J. Polym. Sci. 39 (2021) 1246–1261. doi: 10.1007/s10118-021-2631-y
W. Yuan, X. Qu, Y. Lu, et al., Chin. Chem. Lett. 32 (2021) 2021–2026. doi: 10.1016/j.cclet.2020.12.003
Q.L. Zhu, C. Du, Y. Dai, et al., Nat. commun. 11 (2020) 1–11. doi: 10.1038/s41467-019-13993-7
H. Fan, J.P. Gong, Macromolecules 53 (2020) 2769–2782. doi: 10.1021/acs.macromol.0c00238
J.Y. Sun, X. Zhao, W.R. Illeperuma, et al., Nature 489 (2012) 133–136. doi: 10.1038/nature11409
H. Xu, X.M. Xie, Chin. Chem. Lett. 32 (2021) 521–524. doi: 10.1016/j.cclet.2020.04.039
W.Z. Lian, Z.W. Fan, K. Cui, et al., Macromolecules 54 (2021) 8996–9006. doi: 10.1021/acs.macromol.1c01064
G. Huang, Z. Tang, S. Peng, et al., Macromolecules 55 (2021) 156–165.
H.C. Yu, S.Y. Zheng, L. Fang, et al., Adv. Mater. 32 (2020) 2005171. doi: 10.1002/adma.202005171
Y.J. Wang, X.N. Zhang, Y. Song, et al., Chem. Mater. 31 (2019) 1430–1440. doi: 10.1021/acs.chemmater.8b05262
C.N. Zhu, S.Y. Zheng, H.N. Qiu, et al., Macromolecules 54 (2021) 8052–8066. doi: 10.1021/acs.macromol.1c00835
C. Du, X.N. Zhang, T.L. Sun, et al., Macromolecules 54 (2021) 4313–4325. doi: 10.1021/acs.macromol.1c00152
X.N. Zhang, Y.J. Wang, S. Sun, et al., Macromolecules 51 (2018) 8136–8146. doi: 10.1021/acs.macromol.8b01496
Q.B. Tong, C. Du, Z. Wei, et al., J. Mater. Chem. B 9 (2021) 9863–9873. doi: 10.1039/D1TB02114A
X.P. Hao, C.Y. Li, C.W. Zhang, et al., Adv. Funct. Mater. 31 (2021) 2105481. doi: 10.1002/adfm.202105481
C.N. Zhu, T. Bai, H. Wang, et al., Adv. Mater. 33 (2021) 2102023. doi: 10.1002/adma.202102023
C.N. Zhu, C.Y. Li, H. Wang, et al., Adv. Mater. 33 (2021) 2008057. doi: 10.1002/adma.202008057
H.C. Yu, X.P. Hao, C.W. Zhang, et al., Small 17 (2021) 2103836. doi: 10.1002/smll.202103836
Y. Yao, Z. Huang, P. Xie, et al., Science 359 (2018) 1489–1494. doi: 10.1126/science.aan5412
F.M. Menger, K.D. Gabrielson, Angew. Chem. Int. Ed. 34 (1995) 2091–2106. doi: 10.1002/anie.199520911
D.E. Discher, A. Eisenberg, Science 297 (2002) 967–973. doi: 10.1126/science.1074972
Y. Zhou, D. Yan, J. Am. Chem. Soc. 127 (2005) 10468–10469. doi: 10.1021/ja0505696
Y. Jiang, Y. Wang, Z. Xu, et al., Acc. Mater. Res. 1 (2020) 175–187. doi: 10.1021/accountsmr.0c00027
Y. He, Y. Liu, F. Guo, et al., Chin. Chem. Lett. 31 (2020) 1625–1629. doi: 10.1016/j.cclet.2019.10.010
Q.L. Meng, H. Liu, Z. Huang, et al., Chin. Chem. Lett. 29 (2018) 711–715. doi: 10.1016/j.cclet.2017.10.028
Z. Teng, B. Wang, Y. Hu, et al., Chin. Chem. Lett. 30 (2019) 717–720. doi: 10.1016/j.cclet.2018.08.017
Z. Xu, C. Gao, ACS Nano 5 (2011) 2908–2915. doi: 10.1021/nn200069w
B. Fang, D. Chang, Z. Xu, et al., Adv. Mater. 32 (2020) 1902664. doi: 10.1002/adma.201902664
W.Z. Fang, L. Peng, Y.J. Liu, et al., Chin. J. Polym. Sci. 39 (2021) 267–308. doi: 10.1007/s10118-021-2515-1
P. Ma, P. Li, Y. Wang, et al., Chin. J. Polym. Sci. 39 (2021) 1657–1664. doi: 10.1007/s10118-021-2619-7
Z. Li, Z. Xu, Y. Liu, et al., Nat. commun. 7 (2016) 13684. doi: 10.1038/ncomms13684
X. Zhang, Y. Guo, Y. Liu, et al., Carbon 167 (2020) 249–255. doi: 10.1016/j.carbon.2020.05.051
D. Chang, J. Liu, B. Fang, et al., Science 372 (2021) 614–617. doi: 10.1126/science.abb6640
W.H. Otto, J. Am. Ceram. Soc. 38 (1955) 122–125.
J. Zhang, B. He, Y. Hu, et al., Adv. Mater. 33 (2021) 2008071. doi: 10.1002/adma.202008071
B. Valeur, M.N. Berberan-Santos, J. Chem. Educ. 88 (2011) 731–738. doi: 10.1021/ed100182h
R. Hoffmann, Acc. Chem. Res. 4 (1971) 1–9. doi: 10.1021/ar50037a001
H. Zhang, Z. Zhao, P.R. McGonigal, et al., Mate. Today 32 (2020) 275–292. doi: 10.1016/j.mattod.2019.08.010
D.A. Tomalia, B. Klajnert-Maculewicz, K.A.M. Johnson, et al., Prog. Polym. Sci. 90 (2019) 35–117. doi: 10.1016/j.progpolymsci.2018.09.004
S. Tang, T. Yang, Z. Zhao, et al., Chem. Soc. Rev. 50 (2021) 12616–12655. doi: 10.1039/D0CS01087A
Z. Zhang, W. Yan, D. Dang, et al., Cell. Rep. Phys. Sci. 3 (2022) 100716. doi: 10.1016/j.xcrp.2021.100716
Z. Zhang, Z. Zhang, H. Zhang, et al., J. Polym. Sci. 60 (2022) 2127–2135. doi: 10.1002/pol.20210954
H. Zhang, B.Z. Tang, JACS Au 1 (2021) 1805–1814. doi: 10.1021/jacsau.1c00311
J. Zhang, L. Hu, K. Zhang, et al., J. Am. Chem. Soc. 143 (2021) 9565–9574. doi: 10.1021/jacs.1c03882
X. Jiang, W. Tao, C. Chen, et al., Chem. Sci. 12 (2021) 15928–15934. doi: 10.1039/D1SC05426K
N. Huang, P. Wang, D. Jiang, Nat. Rev. Mater. 1 (2016) 1–19.
N. Huang, K.H. Lee, Y. Yue, et al., Angew. Chem. 132 (2020) 16730–16736.
S. Wang, X. Xu, Y. Yue, et al., Small Structures 1 (2020) 2000021. doi: 10.1002/sstr.202000021
X. Xu, S. Wang, Y. Yue, et al., ACS Appl. Mater. Inter. 12 (2020) 37427–37434. doi: 10.1021/acsami.0c06022
Y. Yue, P. Cai, X. Xu, et al., Angew. Chem. Int. Ed. 60 (2021) 10806–10813. doi: 10.1002/anie.202100717
Y. Yue, P. Cai, K. Xu, et al., J. Am. Chem. Soc. 143 (2021) 18052–18060. doi: 10.1021/jacs.1c06238
Y. Yue, H. Li, H. Chen, et al., J. Am. Chem. Soc. 144 (2022) 2873–2878. doi: 10.1021/jacs.1c13012
J. Hou, O. Inganas, R.H. Friend, et al., Nat. Mater. 17 (2018) 119–128. doi: 10.1038/nmat5063
S. Li, C.Z. Li, M. Shi, et al., ACS Energy Lett. 5 (2020) 1554–1567. doi: 10.1021/acsenergylett.0c00537
Z.P. Yu, K. Yan, W. Ullah, et al., ACS Appl. Polym. Mater. 3 (2021) 60–92. doi: 10.1021/acsapm.0c00791
C. Yang, S. Zhang, J. Ren, et al., Chin. Chem. Lett. 32 (2021) 2274–2278. doi: 10.1016/j.cclet.2021.03.006
M. Luo, C. Zhu, J. Yuan, et al., Chin. Chem. Lett. 30 (2019) 2343–2346. doi: 10.1016/j.cclet.2019.07.023
L. Zuo, S.B. Jo, Y. Li, et al., Nat. Nanotechnol. 17 (2021) 53–60.
X. Li, R. Xia, K. Yan, et al., Chin. Chem. Lett. 31 (2019) 1608–1611.
Z.P. Yu, X. Li, C. He, et al., Chin. Chem. Lett. 31 (2019) 1991–1996.
Z. Zhang, S. Zhang, Z.X. Liu, et al., Acta Phys. Chim. Sin. 35 (2019) 394–400. doi: 10.3866/PKU.WHXB201805091
Z.P. Yu, Z.X. Liu, F.X. Chen, et al., Nat. commun. 10 (2019) 2152. doi: 10.1038/s41467-019-10098-z
S. Li, L. Zhan, F. Liu, et al., Adv. Mater. 30 (2018) 1705208. doi: 10.1002/adma.201705208
R. Qin, W. Yang, S. Li, et al., Mater. Chem. Front. 3 (2019) 513–519. doi: 10.1039/C8QM00609A
T.J. Wen, D. Wang, L. Tao, et al., ACS Appl. Mater. Interfaces 12 (2020) 39515–39523. doi: 10.1021/acsami.0c12100
S. Li, L. Zhan, C. Sun, et al., J. Am. Chem. Soc. 141 (2019) 3073–3082. doi: 10.1021/jacs.8b12126
S. Li, L. Zhan, N. Yao, et al., Nat. commun. 12 (2021) 4627. doi: 10.1038/s41467-021-24937-5
T.J. Wen, Z.X. Liu, Z. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 12964–12970. doi: 10.1002/anie.202101867
Z.X. Liu, Z.P. Yu, Z. Shen, et al., Nat. commun. 12 (2021) 3049. doi: 10.1038/s41467-021-23389-1
X. Chen, D. Wang, Z. Wang, et al., Chem. Eng. J. 424 (2021) 130397. doi: 10.1016/j.cej.2021.130397
Q.Q. Zhang, Y. Li, D. Wang, et al., Bull. Chem. Soc. Jpn. 94 (2020) 183–190.
Y. Li, X. Huang, K. Ding, et al., Nat. commun. 12 (2021) 1–9. doi: 10.1038/s41467-020-20314-w
D. Wang, G. Zhou, Y. Li, et al., Adv. Funct. Mater. 32 (2021) 2107827.
D. Wang, H. Liu, Y. Li, et al., Joule 5 (2021) 945–957. doi: 10.1016/j.joule.2021.02.010
M.A. Green, E.D. Dunlop, J. Hohl-Ebinger, et al., Prog. Photovolt. Res. Appl. 29 (2021) 3–15. doi: 10.1002/pip.3371
Y. Li, C. He, L. Zuo, et al., Adv. Energy Mater. 11 (2021) 2003408. doi: 10.1002/aenm.202003408
S. Ye, S. Chen, S. Li, et al., ChemSusChem 14 (2021) 3599–3606. doi: 10.1002/cssc.202100689
Y. Pan, X. Zheng, J. Guo, et al., Adv. Funct. Mater. 32 (2022) 2108614. doi: 10.1002/adfm.202108614
S. Wang, Y. Tao, S. Li, et al., Macromolecules 54 (2021) 7862–7869. doi: 10.1021/acs.macromol.1c01301
Y. Yu, W. Tao, L. Wang, et al., J. Mater. Chem. A 9 (2021) 22926–22933. doi: 10.1039/D1TA06416A
L. Zhan, S. Li, X. Xia, et al., Adv. Mater. 33 (2021) 2007231. doi: 10.1002/adma.202007231
J. Huang, D. Zhao, Z. Dou, et al., Energy Environ. Sci. 14 (2021) 3010–3018. doi: 10.1039/D0EE03387A
A. Sakdinawat, D. Attwood, Nat. Photonics 4 (2010) 840–848. doi: 10.1038/nphoton.2010.267
W.C. Pan, H.D. Wu, J.J. Luo, et al., Nat. Photonics 11 (2017) 726. doi: 10.1038/s41566-017-0012-4
Q. Dong, Y. Fang, Y. Shao, et al., Science 347 (2015) 967–970. doi: 10.1126/science.aaa5760
B. Peng, R. Wu, H. Li, Acc. Chem. Res. 54 (2021) 4498–4507. doi: 10.1021/acs.accounts.1c00537
W. Wei, Y. Zhang, Q. Xu, et al., Nat. Photonics 11 (2017) 315. doi: 10.1038/nphoton.2017.43
G.N. Ankah, P. Buchele, K. Poulsen, et al., Org. Electron. 33 (2016) 201–206. doi: 10.1016/j.orgel.2016.03.023
G. Yu, J. Gao, J.C. Hummelen, et al., Science 270 (1995) 1789–1791. doi: 10.1126/science.270.5243.1789
Q. Wen, W. Ma, Y. Liu, et al., J. Phys. Chem. Lett. 12 (2021) 11176–11181. doi: 10.1021/acs.jpclett.1c03297
J. Ren, M. Niu, X. Guo, et al., J. Am. Chem. Soc. 142 (2020) 1630–1635. doi: 10.1021/jacs.9b13087
H. Li, H.L. Xin, D.A. Muller, et al., Science 326 (2009) 1244–1247. doi: 10.1126/science.1178583
J. Ren, Y. Liu, H. Li, J. Polym. Sci. 60 (2021) 1151–1173.
T.E. Schlesinger, J.E. Toney, H. Yoon, et al., Mat. Sci. Eng. R. 32 (2001) 103–189. doi: 10.1016/S0927-796X(01)00027-4
J. Androulakis, S.C. Peter, H. Li, et al., Adv. Mater. 23 (2011) 4163–4167. doi: 10.1002/adma.201102450

Figure 1 Versatile organoboron catalysts for ring-opening (co)polymerizations. Reproduced with permission [2]. Copyright 2021, American Chemical Society.

Figure 2 (a) Mechanism diagram of CFP using excess anhydride-formable dicarboxylic acid and primary diol as monomers to afford HMW polyesters. (b) Effect of monomer structure on molecular weight and diacid/diol composition ratio (R/R') of polyesters via CFP. The initial feed ratio of diacid/diol monomers was 1.1:1. Reproduced with permission [28]. Copyright 2021, Elsevier.

Figure 3 (a) Proposed mechanism for the formation of amino-quinone networks that integrate the displacement of interfacial water by amino groups and the subsequent cross-linking via covalent reactions. (b) Molecular structures of 5-hydroxy-1,4-naphthoquinone (HNQ) and polyamine (PEI). (c) Water contact angles of different planar substrates before and after HNQ-PEI coating. (d) Treatment capacity of a series of dye solutions. (e-i) Schematic of liquid-phase epitaxy of CC3 organic cages (e), chelation of metal ions (f), mineralization of FeOOH nanorods (g), metallization of Ag nanoparticles (h), and self-assembly of monolayers (i) on the coating surface. Reproduced with permission [41]. Copyright 2021. Wiley-VCH.

Figure 4 (a) TEB-TEA pair-catalyzed PO-CO2 copolymerization via a zwitterionic mechanism. Reproduced with permission [62]. Copyright 2021, American Chemical Society. (b) DMCC-catalyzed IBO-cyclic anhydride copolymerization to form semicrystalline polyesters. Reproduced with permission [66]. Copyright 2021, American Chemical Society. (c) Altering chain flexibility of aliphatic polyesters for yellow-green clusteroluminescence in 38% quantum yield. Reproduced with permission [72]. Copyright 2021, Wiley-VCH.

Figure 5 The fabrication supramolecular prodrug nano-assemblies (SPNAs) for RNS-potentiated chemotherapy. (A) The intracellular generation of ONOO− by the co-delivery of CDDP and NO. (B) Schematic illustration of SPNAPt/NO for ONOO− potentiated Pt-based chemotherapy. Reproduced with permission [88]. Copyright 2021, American Chemical Society.

Figure 6 (a) Synthesis and chemical structure of fluorescent shape-memory hydrogel. (b) Temperature-mediated shape-memory effect and photo-induced tunable fluorescence of the hydrogel. Photos to show the decryption process of architected hydrogels by employing origami (c) and kirigami technologies (d). The hydrogel recovered to flat shape upon heating, and the encrypted information became readable under UV light irradiation. Scale bar: 1 cm. Reproduced with permission [107]. Copyright 2021, Wiley-VCH.

Figure 7 (a) Schematic of precisely reversible fusion and fission of multiple GO fibers. (b, c) Scanning electron microscopy images showing the water-induced fusion and fission processes of 100 GO fibers. Reproduced with permission [124]. Copyright 2021, American Association for the Advancement of Science (AAAS).

Figure 8 (a) The role of amide (n,π*) transitions in polypeptide clusteroluminescence. Reproduced with permission [132]. Copyright 2022, Elsevier. (b) The mysterious blue emission around 440 nm in carbonyl-based aliphatic clusteroluminogens. Reproduced with permission [133]. Copyright 2022, Wiley-VCH. (c) Through-space interactions in clusteroluminescence. Reproduced with permission [134]. Copyright 2021, CC-BY.Conductive covalent organic frameworks

Figure 9 (a) Chemical structure and (b) schematic representation of tetraaza[14]annulene-linked NiPc-CoTAA COF from top view. Reproduced with permission [141]. Copyright 2021, Wiley-VCH. (c) Chemical structure and (d) schematic representation of dioxin-linked CuPcF8-CoPc-COF from top view. Reproduced with permission [142]. Copyright 2021, American Chemical Society. (e) Chemical structure and (f) schematic diagram of piperazine-linked NiPc-NH-CoPcF8 from top view. Reproduced with permission [143]. Copyright 2022, American Chemical Society.

Figure 10 (a) Schematic diagram of the non-fused ring electron acceptors (NFRAs). Reproduced with permission [159]. Copyright 2021, Wiley-VCH. (b) The plausible photo-isomerization and reaction mechanism of the vinyl group in A-D-A NFAs, and the structural factors from the molecular level to macroscopic condensed solid for enhancing photostabilities of NFAs. Reproduced with permission [160]. Copyright 2021, CC-BY. (c) Schematic diagram to illustrate the fabrication of eco-friendly PSCs for agrivoltaics. Reproduced with permission [164,165]. Copyright 2021, Elsevier and 2022, Wiley-VCH.

Figure 11 (a) Schematic of PbI2 crystallization in a TiO2 gel and a resulting gel-grown crystal with an interpenetrating gel network inside. (b) OM images of a gel-grown PbI2 crystal. (c) Single-crystal XRD pattern of a gel-grown PbI2 crystal. (d) XPS spectra (Ti 2p signal) of a gel-grown PbI2 crystal without and with partially etching. (e) X-ray response photocurrent density as a function of dose rate at a bias of 15 V. (f, g) PL and TRPL spectra, respectively. (h) Illustration of the PbI2-TiO2 BHJs direct X-ray detector. (i) Bias-dependent steady-state photocurrent density under a 450 nm laser. (j) Schematic illustration of the X-ray exciton generation-separation process. Reproduced with permission [185]. Copyright 2021, American Chemical Society.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: