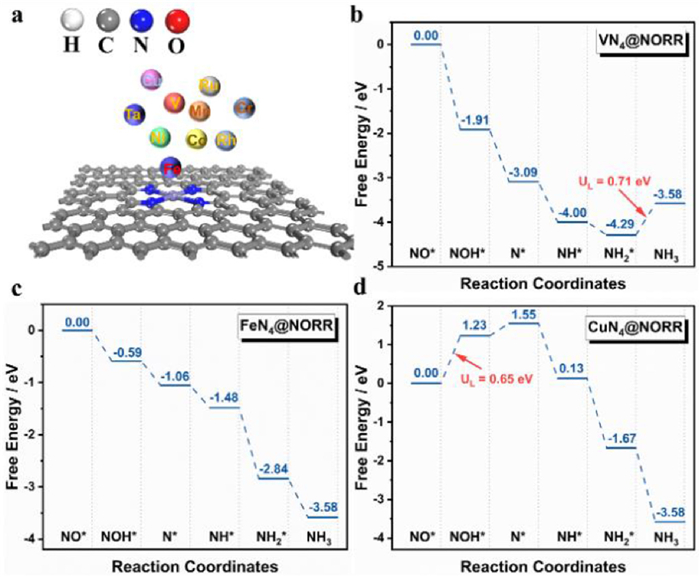
Figure 1.
(a) Structure of TM-N4 and the considered TM atoms. (b-d) Free energy diagrams of NO reduction to NH3 on V-N4, Fe-N4 and Cu-N4 under 0 V vs. RHE.
Electroreduction NO to NH3 over single metal atom anchored on pyrrole type defective graphene: A DFT study
Jiazhi Wang , Kai Li , Qi Hao , Dongxue Liu , Xinbo Zhang

Nowadays, traditional fossil fuel burning provides the main energy for development, while it also brings a large emission of pollutants. Nitric oxide (NO), one of the major air pollutants, will cause serious environmental problems, such as acid rain, photo-chemical smog, and ozone depletion [1,2]. Previous studies have been focused on the selective catalytic reduction (SCR) of NO to N2 and H2O using NH3, which has been applied industrially for decades [3-5]. However, the process of SCR technology has been still environmentally polluting and operated costly.
Ammonia, as an important chemical raw material, is mainly synthesized by the industrial Haber-Bosch process, which converts N2 into NH3 under high temperature and high pressure [6]. The Haber-Bosch process also consumes a lot of resources and causes environmental pollution. In recent years, many researchers have devoted themselves to finding a suitable method to replace the Haber-Bosch process for NH3 synthesis. The electrochemical reduction of N2 to NH3 is considered to be a reasonable alternative due to its high efficiency, low cost, and environmental friendliness [7-9]. Although a number of works on eletrochemical NH3 synthesis have been published and some progress has been made, the low selectivity and sluggish reaction kinetics of NRR hinder its development, which is attributed to the fact that the N≡N triple bond is too strong to break. Fortunately, the N—O bond strength of NO is lower than the N≡N triple bond of N2, and the reduction of NO is relatively easy. Thus, the direct electrochemical reduction of NO to NH3 can not only solve the environmental pollution caused by NO but also generate NH3 for industrial production. To date, few reports are reported for NO electroreduction to NH3 (NORR) [10-12]; for example, Sun et al. reported the use of MoS2 nanosheet on graphite felt (MoS2/GF) as an efficient and robust electrocatalyst for the conversion of NO to NH3 with a maximum faraday efficiency of 76.6% and an NH3 yield of 99.6 µmol cm−2 h−1 [10]. The efficiency of NORR is highly dependent on the catalyst; therefore, exploring or designing an efficient catalyst is the key to improving the development of NORR.
Up to now, many works report that single-atom catalysts (SACs) can be employed as the effective catalyst in electrocatalysis, such as hydrogen evolution reaction (HER) [13], oxygen evolution/reduction reaction (ORR/OER) [14,15] and CO2 reduction reaction (CO2RR) [16,17], which are attributed to its high efficiency of metal atom utilization and excellent catalytic activity. For SACs, the support is very important, which can effectively make metal atoms high dispersion and realize their high activity. In addition, in the electrochemical reaction, the support must be conductivity and acid/alkaline resistance. Graphene, as a two-dimensional (2D) material with excellent physical and chemical properties, has been widely employed as support for anchoring the metal atoms and producing the SACs [18-20]. Among the graphene-based SACs, the transition metals anchored on pyrrole-type N-doped graphene single atom electrocatalyst (TM-N4) is considered to be one of the most effective structures for electrochemical reaction [21,22]. Previous experimental and theoretical studies have demonstrated that TM-N4 shows high activity for ORR/OER; for example, Hu et al. successfully prepared catalysts with pyrrole-type Mn-N4 single atom, which exhibited outstanding ORR activity in alkaline, acidic and neutral electrolytes [21]. Therefore, the pyrrole-type TM-N4 can serve as a promising catalyst for the NORR process.
For the electrochemical reaction of ORR, Nørskov et al. have proposed a volcanic relationship of activity, in which the free adsorption energy of OH species can be acted as a descriptor for reaction activity [23]. Based on such a relationship, the catalyst with excellent activity can be predicted and designed. However, few theoretical studies have systematically explored the activity relationship in NORR, which is expected to design a potential catalyst.
In this study, we employed DFT calculations to screen the promising TM-N4 (TM = V, Cr, Mn, Fe, Co, Ni, Cu, Ru, Rh, Ta) catalysts for NO electroreduction to NH3. It was found that Fe-N4 can sufficiently adsorb and activate the NO molecule. Meanwhile, a high NORR activity with working potential of 0.58 V is obtained. Furthermore, the suppressed HER on Fe-N4 has an excellent selection with a high faraday efficiency of NO electroreduction to NH3. Moreover, our work demonstrated that the free adsorption energy of N (ΔGads(*N)) on different metal centers shows a linear relationship with the activity of NORR. Based on these results, we have proposed a promising strategy to design the rational catalysts for electrochemical NO removal and NH3 synthesis.
For single atoms, the basic criterion for designing single atoms is preventing atomic aggregation, thus, the value of binding energy (Eb) should be more negative compared to cohesive energy (Ec) to avoid the aggregation of atoms. The binding energies for TM-N4 are calculated as Eb = E(TM-N4) – ETM – Eatom, where E(TM-N4), ETM and Eatom refer to the total energies of TM-N4, pristine N-doped graphene, and isolated atom in the vacuum, respectively. Whereas the Ec for the TM atoms is defined as Ec = (Ebulk – nEatom)/n, where Ebulk is the energy of bulk metal, and n is the number of metal atoms in the crystal. The calculated results are listed in Table S1 (Supporting information), and the value indicated that the transition atoms prefer to form a single atom on the pyrrole-type N-doped graphene. What is more, the metal atom has a lot of electrons transferred to N atoms around the metal atom, implying that the metal atom and the four surrounding N atoms also have strong interactions to keep the metal atom stable based on the barde charge analysis, shown in Table S2 (Supporting information).
For electroreduction of NO to NH3, there are two possible pathways, i.e., path 1: NO → N + O → NH3 + H2O (dissociation pathway) and path 2: NO → NOH/HNO → NH3 + H2O (associative pathway, similar to NRR process). For path 1, the N—O bond will be broken first to generate isolated N* and O* absorbed on the surface, and then the resulting N* and O* can be protonated to form NH3 and H2O, respectively. Since the N—O bond is too strong to be broken and directly dissociated on the catalytic surface, thus, path 1 is not favorable (Fig. S17 in Supporting information). In path 2, the adsorbed NO will be hydrogenated into NOH* or HNO* first. Since the H atom can supply electrons to NO, the N—O bond will weaken. As the hydrogenation process proceeds, the N—O will become weaker and finally be broken to form NH3 and H2O. Therefore, in our work, path 2 will be considered the main channel for NORR. Since the metal atom is usually acted as the center for most reactions [24], it will be considered the main active site for NORR.
For NORR, the effective adsorption of NO on the catalyst plays an important role in the subsequent reaction [25,26], and Fig. 1a shows the different transition metals anchored on pyrrole type defective of graphene with TM-N4 structure for NORR. There are three possible adsorption structures for NO, i.e., the N end-on, the O end-on, and side-on adsorption, respectively shown in Fig. S1 (Supporting information). The side-on configuration of NO on the surface is unstable after structure optimization, and the NO is tended to adsorb on the surface by N end-on configuration compared to the adsorption energy of N end-on (−2.04 eV) with O end-on (−0.64 eV) configuration. Thus, the N end-on adsorption structure will be discussed without considering the side-on structure in the following text.
For the 3d transition metal system (V, Cr, Mn, Fe, Co, Ni and Cu), it was found that the adsorption capacity of NO showed a decreasing trend which may be attributed to the occupied electron of the 3d orbital increases; the adsorption of NO on the TM-N4 is shown in Fig. S2 (Supporting information), in which the catalyst V-N4 shows the strongest adsorption of NO (∆Eads = −2.76 eV), while the Cu-N4 shows the weakest adsorption of NO (∆Eads = −0.31 eV). Moreover, the same trend is also observed in the 4d and 5d transition metal systems (Ru, Rh and Ta), in which the Ta-N4 shows the strongest adsorption of NO (∆Eads = −4.04 eV). It is also noted that the average adsorption of NO on 4d and 5d is stronger than that of the 3d system, indicating that the interaction of the 4d (or 5d) orbital with the 2p orbital of NO is stronger than 3d-2p of 3d metal atoms. This is well in agreement with that the 3d orbital is more local than that of the 4d and 5d orbital. Overall, transition metal atoms with less occupied d electron and local d orbital, such as V-N4 and Ta-N4 catalysts, will show high affinity for NO. And those with almost filled d orbital or local d orbital, such as CuN4, cannot effectively adsorb NO gas. Thus, the Cu-N4 system exhibits lower activity toward NORR as shown in Fig. 1d. In addition, the differential charge for NO adsorption on TM-N4 is shown in Fig. S3 (Supporting information). It is seen that the electron will be transferred from catalysts to adsorb NO species, indicating that the *NO will be activated and the following reaction can occur.
For the N end-on adsorption structure, there are four possible hydrogenation pathways, i.e., O-distal, N-distal, O-alternative, and N-alternative shown in Fig. S4 (Supporting information). In O-distal (or N-distal) pathway, the O (or N) atom of NO will be firstly hydrogenated to H2O (or NH3), then the other atom begins to be reduced. In O-alternative (or N-alternative) pathway, O and N are alternately hydrogenated [27,28]. The above four mechanistic reactions are summarized in Table S3 (Supporting information). According to previous literature [28], the end-on adsorption of NO is more favorable to go through the O-distal pathway than the N-distal pathway. Thus, in this work, the O-distal pathway is served as the main pathway for NORR over TM-N4.
To understand the whole process better, the free energy changes (∆G) for the electroreduction of NO to NH3 through the O-distal pathway have been investigated. The free energy curves of V-N4, Fe-N4, and Cu-N4 are shown in Figs. 1b–d, while others are shown in Figs. S5−S11 (Supporting information). The corresponding structures of the reaction intermediates and adsorption data are shown in Fig. S12 and Table S4 (Supporting information). For NO* → NOH*, it is seen that Ni-N4 (0.65 eV, Fig. S6) and Cu-N4 (1.23 eV, Fig. 1d) show the endothermic process, while the corresponding step is exothermic processes over other TM-N4 catalysts, especially for V-N4 (−1.91 eV, Fig. 1b) and Ta-N4 (−3.15 eV, Fig. S9) with the highest downhill ∆G. It is interesting that the adsorption energy of NO has a linear relationship with ∆G(*NOH) (Fig. S13 in Supporting information), indicating that the high adsorption of NO can improve the *NO activation. Thus, the weak affinity for NO on Cu-N4 and Ni-N4 makes the NO* → NOH* become the potential-determining step (PDS), and these two catalysts will show low activity for NORR. Moreover, a similar ∆G trend is also found for NOH* → N*, i.e., the Cu-N4 and Ni-N4 show an endothermic process, while the other configurations show the exothermic process (Figs. 1b–d and Figs. S5−S11). This result further indicates that the weak adsorption of NO on Cu-N4 and Ni-N4 will limit NO hydrogenation to NOH* and N*, which is in agreement with our previous conclusion that the adsorption of NO is the key to NORR.
For the subsequent elementary reaction steps (N* → NH3*), the investigated TM-N4 shows continuous downhill the ∆G, except for Cr-N4, Mn-N4, Ta-N4, V-N4, and Ru-N4. For TM-N4 (TM = Mn and Cr), it is seen that the N* → NH* is an endothermic process and the potential-determining step (Figs. S10 and S11). And for Ta-N4 (Fig. S9), V-N4 (Fig. 1b) and Ru-N4 (Fig. S7), the NH2* → NH3 is the rate-determining step. This suggests that the TM-N4 with strong adsorption of NO will bring the strong adsorption of NH2* and make the desorption of NH3 unfavorable. Thus, based on the discussion above, it can be concluded that the over weak or strong adsorption of NO are both harmful for NORR, which will be limited by the NO activation and NH3 desorption, respectively. Moreover, the TM-N4 (TM = Co, Fe and Rh) with optimized NO adsorption (−1.62, −2.04 and −2.18 eV, respectively) and optimized N* adsorption (2.83, 1.90, 2.38 eV, respectively) show potential activity for NORR.
In NORR, ∆G of elementary reaction (protonation) is affected by the external potential, i.e., as the external increases, the ∆G will be less exothermic (usually used in ORR). When one of the elementary reactions becomes uphill, the NORR activity will be hindered. Thus, the elementary reaction with the least exothermic will affect the performance. It is seen that there exists the highest potential (noted as working potential), which can keep all the elementary reactions exothermic. The higher the working potential, the higher activity will be obtained. For TM-N4 (TM = Co, Fe, and Rh), the elementary step of NO* → NOH* with ∆G is −0.02, −0.58 and −0.36 eV, respectively. It is clear that the Fe-N4 catalyst with an outstanding working potential of 0.58 V shows the highest activity for NORR among the TM-N4 catalysts. However, such a result is based on the elementary reaction of NO* → NOH*. To exclude the influence of side reaction (NO* → HNO*), the dynamic process (the reaction barrier) of two possible reactions is investigated on Fe-N4, and the results are shown in Fig. 2a. IS, TS, FS represent the initial structure, transition structure and final structure for the formation of NOH* and HNO* intermediate, respectively. It is seen that the H adsorbed on C will move to the *NO and interact with N or O to form HNO* and NOH*, respectively. Fig. 2c shows the energy barriers for producing HNO* and NOH* are 1.17 and 0.54 eV, respectively. Thus, the NO* is preferred to be hydrogenated to *NOH over Fe-N4.
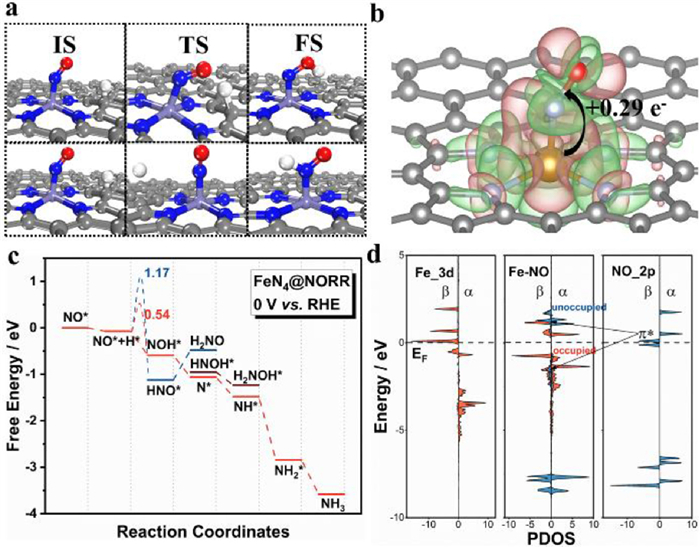
During the NORR process, the competition reaction HER will consume H+/e− and decrease the faraday efficiency. Thus, the HER of Fe-N4 is investigated, and the result is shown in Fig. S14 (Supporting information). It is found that the activity of HER is 0.74 eV over Fe-N4, which is higher than the adsorption of free energy of NO* (−2.04 eV). This indicates that NO adsorption on Fe-N4 will be dominant, and NORR can occur without interference. Therefore, the Fe-N4 will show excellent selectivity with high faraday efficiency during NORR.
According to our previous discussion, the optimized NO* is the beginning of the high activity of NORR. Thus, in this part, we will deeply analyze the activation mechanism of NO* over Fe-N4. From the differential charge of NO* on Fe-N4 (Fig. 2b), the adsorbed NO* obtains ~0.29 electrons from support, and the accumulated charge on NO* will promote the reduction reaction. The differential charge density also suggests that the adsorbed NO* can interact with the Fe-N4 by the so-called "push−pull" hypothesis, in which Fe-N4 can "push" electrons into the anti-bonding orbitals of NO and simultaneously "pull" the lone-pair electrons from the NO, which is confirmed by the density of states (DOS).
To elucidate the binding nature of the species involved in O-distal mechanism, the partial density of states (PDOS) of Fe-N4 before and after NO adsorption were analyzed to further reveal the interaction of Fe atom and NO. In Fig. 2d, it is seen that both unoccupied and occupied Fe-3d orbitals of Fe-N4 contribute to NO* activation. During NO adsorption, the π* orbitals of NO molecules interact with the Fe-3d orbitals to form partially occupied d-π* orbitals. Meanwhile, occupied d-π* orbitals are observed due to the hybridization between the occupied Fe-d orbitals and NO-σ orbitals below the Fermi level (EF). Therefore, the mechanism of the interaction between Fe and NO can be attributed to the "donation/back" of electrons. On the one hand, the empty TM-d orbital can accept electrons from the NO-σ orbital. On the other hand, the occupied TM-d orbitals can donate electrons back to the empty π* orbitals of NO molecules. This "donate/ back" mechanism can also be verified on other metals [29].
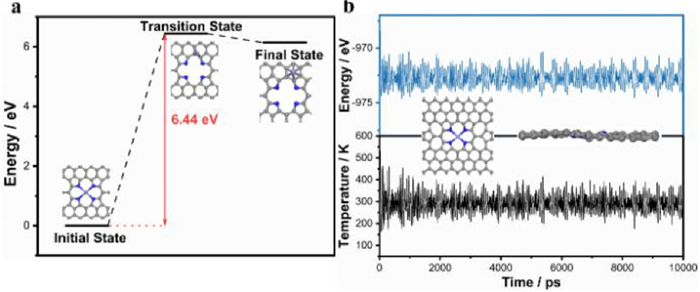
During the NORR, the Fe atom in Fe-N4 will be occupied by the intermediate, which makes Fe be oxidation. Therefore, the evaluation of the stability of Fe-N4 is very important for NORR. The two decorations of Fe atom on different sites of N-doped carbon are investigated. One is the Fe atom anchored on the pyrrole-type hole that interacts with four N atoms (A); the other is the Fe atom in the hollow site of a carbon six-member ring bonding to six coordinated C atoms (B) (Fig. 3a). We find that the formation of the B-structure from the A-structure requires the energy of 6.44 eV to overcome the barrier, indicating that the four N atoms around the Fe atom are binding the Fe atom strongly and make the Fe atom break out difficultly to form other structures, which also indicates that the structure is relatively stable. In addition, such a barrier is also higher than the adsorption energy of all the intermediate, suggesting the adsorption of intermediate cannot make Fe out of the surface. Moreover, ab initio molecular dynamics (AIMD) simulations are performed to prove the kinetic stability of Fe-N4. The potential energy and temperature oscillate near the initial condition (Fig. 3b), and the geometric structure of Fe-N4 remains integrity (insert in Fig. 3b) after 300 K for 10 ps. Overall, Fe-N4 can serve as efficient NORR catalysts for selective ammonia synthesis with high stability and activity.
Nørskov et al. have successfully proposed a scale relationship between the adsorption energies of intermediates in electro-chemistry reactions. And a valid descriptor can be proposed based on this scaling relationship, which can be used to efficiently screen and predict promising catalysts for the reaction. Therefore, aimed to explore a descriptor for NORR, we try to conclude all the adsorption energies of all intermediates involved O-distal pathway for NORR on ten (TM-N4) catalysts; the details are shown in Fig. S15 (Supporting information).
Interestingly, the adsorption free energies of intermediates in the calculated TM-N4 showed good scale with respect to the adsorption free energy of N* for the O-distal pathway. It can be seen from Fig. S15 that there is a good correlation coefficient (R2 close to 0.90) scale relationship between GNOH*, GNH*, GNH2* and GN* on the 10 TM-N4 catalysts, implying that GN* can be used as a descriptor to describe the relationship between the intermediates involved in the O-distal pathway. This can also be reflected by the adsorption structures, in which all through intermediate is through N atom to interact with TM atoms. Thus, the adsorption of the intermediate trend can be directly described by the adoption configuration of N. The scale relationships are shown below:
|
|
(1) |
|
|
(2) |
|
|
(3) |
It is seen that the slope for Eq. 2 is 0.86. This slope is closer to 1 than the other two scale relationship, indicating that the free energy changes for N* → NH* are not sensitive on the investigated TM-N4. Thus, we chose the NO → NOH* and NH2* → NH3 as the basis for drawing the volcano activity scaling.
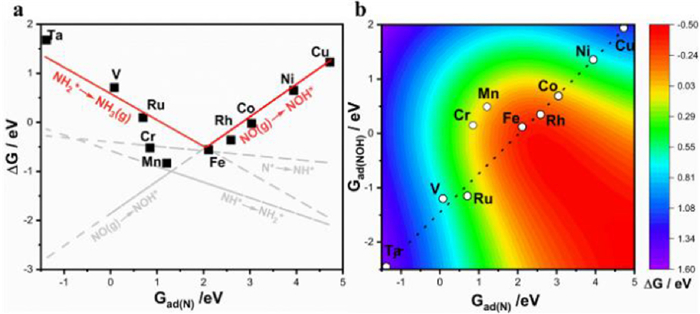
Based on the above scaling relations, the adsorption free energy of the N* (Gads(N*)) can be chosen as the descriptor to predict and screen the catalytic performance of single-atom catalysts for the NORR. And we can establish the relationship between the free energy change ∆G of each reaction step with that of Gads(N*), showed in Fig. 4a. According to our previous discussion, the ∆G with the least exothermic or the largest endothermic will be the PDS for the NORR. In Fig. 4a, the lines for ∆G of NO → NOH* and NH2* → NH3 are over the others; therefore, they are the limit lines for the NORR activity. It is seen that both two lines have an intersection at Gads(N*) = 2.05 eV. When the Gads(N*) < 2.05 eV, the TM-N4 will show strong adsorption for N, and the PDS is the desorption of NH3 (NH2* → NH3). On the contrary, the weak adsorption of N and PDS of *NO → *NOH is obtained when the Gads(N*) > 2.05 eV. In our investigated catalysts, Fe with the optimized N adsorption energy, whose value is 2.11 eV closing to 2.05 eV, shows the highest activity for NORR. On the basis of these results, we then constructed the color contour plots of the ΔG(PDS) as a function of the adsorption free energy for N* (Gads(N*)) and NOH* (Gads(NOH*)) in Fig. 4b. The chemical reaction is always limited by the scaling relation of adsorption energies, and the Fe-N4 is very close to the ideal region, which also indicates that the Fe-N4 is the best catalyst for NORR in these 10 TM-N4 catalysts. However, the activity area covers over half the area, suggesting that there are more available TM-N4 catalysts with potential activity for NORR.
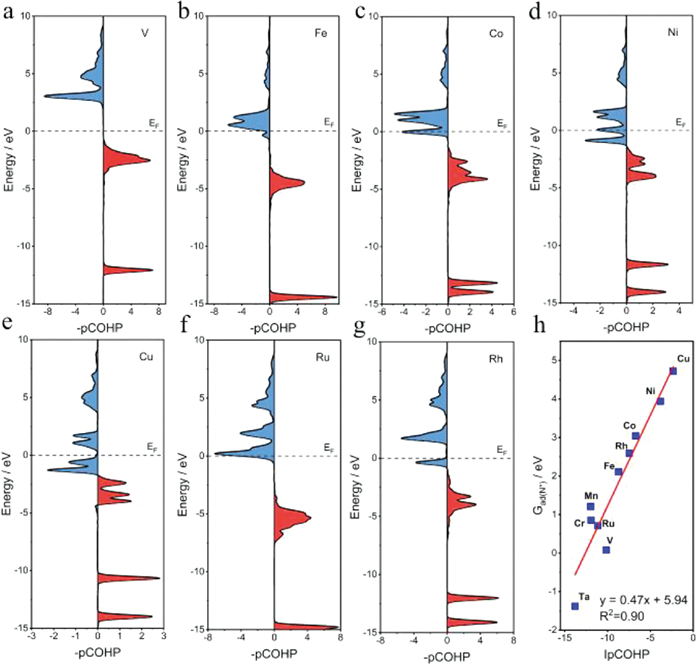
To provide a physical insight into the different activity of TM-N4, the electronic structure of TM-N4 will be investigated. Since the NORR reaction is a multivariate dynamic process, the effective physical factor must be found to describe the reaction activity. According to our previous discussion, the Gads(N*) are related to the NORR activity of TM-N4 catalysts. To reveal the role of different metal centers, the various electronic structures of these TM-N4 catalysts have been analyzed. The poor relationship between the d-band center and Gads(N*) displayed that the d-band center theory is inapplicable for this system because of the existence of discrete states in both the d and sp bands of metals from Fig. S16 (Supporting information), which are different from the pure metals [30]. Thus, we introduced the projected crystal orbital Hamiltonian populations (pCOHP) to describe the relationship between the bond strength of N-TM and Gads(N*), and the results are shown in Fig. 5 and Fig. S18 (Supporting information). For V-N4 (Fig. 5a), all the bonding states are below the Fermi level (EF), while the antibonding orbital group is completely above EF. This means the strong adsorption of N* on V-N4. Meanwhile, for elements from other TM-N4, as shown in Figs. 5b–g, the antibonding orbital gradually moves below the EF, which is consistent with the decrease in bonding strength, as shown by the more aggressive Gads(N*). In addition, we calculated the integrated COHP (ICOHP) by calculating the energy integral to the EF, which directly gives quantitative information. As shown in Fig. 5h, there is a linear relationship between ICOHP and Gads(N*). This suggests that the activity of NORR can be described by the bond strength of N-TM-N4. Thus, the electronic structure of TM affecting the bond strength of N-TM will be the key to exploring the physical nature of NORR activity. The theoretical evaluations of NORR catalysts with excellent activity, stability, and selectivity over TM-N4 provide an ideal pathway for experimental examinations.
In summary, we have investigated TM-N4 electrocatalysts for the direct electrochemical reduction of NO into NH3 through DFT calculation. Our study revealed that Fe-N4, Co-N4 and Rh-N4 have good activity towards NORR, especially Fe-N4 catalyst has excellent selective and working potential. Moreover, we proposed N* intermediate adsorption energy as a descriptor to predict and screen the optimal performance catalysts. In addition, to evaluate the internal of NORR activity over TM-N4, COHP and ICOHP was used to explore the correlation between catalysts and adsorption intermediates N*, suggesting that a moderate N* intermediate adsorption energy is an important indicator for improving NORR activity. Our work provides a guidance for the rational design of NORR catalysts.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the National Natural Science Foundation of China (Nos. 21725103, 52072362), National Key R&D Program of China (No. 2021YFB4000401), and Youth Innovation Promotion Association CAS (No. E1202002).
Supplementary material associated with this article can be found, in the online version, at doi:
K. Li, D.J. Jacob, H. Liao, et al., Nat. Geosci. 12 (2019) 906–910. doi: 10.1038/s41561-019-0464-x
H. He, Y. Wang, Q. Ma, et al., Sci. Rep. 4 (2014) 4172. doi: 10.1038/srep04172
Z. Liu, S. Ihl Woo, Catal. Rev. 48 (2006) 43–89. doi: 10.1080/01614940500439891
C. Liu, J.W. Shi, C. Gao, C. Niu, Appl. Catal. A 522 (2016) 54–69. doi: 10.1016/j.apcata.2016.04.023
L. Han, S. Cai, M. Gao, et al., Chem. Rev. 19 (2019) 10916–10976. doi: 10.1021/acs.chemrev.9b00202
J.H. Montoya, C. Tsai, A. Vojvodic, J.K. Nørskov, ChemSusChem 8 (2015) 2180–2186. doi: 10.1002/cssc.201500322
C. Ling, X. Niu, Q. Li, A. Du, J. Wang, J. Am. Chem. Soc. 140 (2018) 14161–14168. doi: 10.1021/jacs.8b07472
K. Chu, Y. Luo, P. Shen, et al., Adv. Energy Mater. 12 (2022) 2103022. doi: 10.1002/aenm.202103022
Y. Yang, Y. Wang, X. Wang, et al., Adv. Mater. Interfaces 9 (2022) 2101842. doi: 10.1002/admi.202101842
L. Zhang, J. Liang, Y. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 25263–25268. doi: 10.1002/anie.202110879
Z. Wang, J. Zhao, J. Wang, C.R. Cabrera, Z. Chen, J. Mater. Chem. A 6 (2018) 7547–7556. doi: 10.1039/C8TA00875B
H. Niu, Z. Zhang, X. Wang, et al., Small 17 (2021) e2102396. doi: 10.1002/smll.202102396
H. Fei, J. Dong, M.J. Arellano-Jimenez, et al., Nat. Commun. 6 (2015) 8668. doi: 10.1038/ncomms9668
T. Mineva, I. Matanovic, P. Atanassov, et al., ACS Catal. 9 (2019) 9359–9371. doi: 10.1021/acscatal.9b02586
I. Vinogradov, S. Singh, H. Lyle, et al., Nat. Mater. 21 (2022) 88–94. doi: 10.1038/s41563-021-01118-9
S. Chen, W.H. Li, W. Jiang, et al., Angew. Chem. Int. Ed. 61 (2022) e202114450.
M. Li, H. Wang, W. Luo, et al., Adv. Mater. 32 (2020) e2001848. doi: 10.1002/adma.202001848
L. Zhong, L. Zhang, S. Li, ACS Mater. Lett. 3 (2020) 110–120.
S. Ding, Z. Lyu, H. Zhong, et al., Small 17 (2021) e2004454. doi: 10.1002/smll.202004454
L. Zong, K. Fan, W. Wu, et al., Adv. Funct. Mater. 31 (2021) 2104864. doi: 10.1002/adfm.202104864
J. Li, Y.F. Jiang, Q. Wang, et al., Nat. Commun. 12 (2021) 6806. doi: 10.1038/s41467-021-27143-5
L. Yan, L. Xie, X.L. Wu, et al., Carbon Energy 3 (2021) 856–865. doi: 10.1002/cey2.135
R. Christensen, H.A. Hansen, C.F. Dickens, J.K. Nørskov, T. Vegge, J. Phys. Chem. C 120 (2016) 24910–24916. doi: 10.1021/acs.jpcc.6b09141
J. Zhao, Z. Chen, J. Am. Chem. Soc. 139 (2017) 12480–12487. doi: 10.1021/jacs.7b05213
W. Yang, Z. Gao, X. Liu, et al., Fuel 243 (2019) 262–270. doi: 10.1016/j.fuel.2019.01.125
Q. Wu, W. Wei, X. Lv, et al., J. Phys. Chem. C 123 (2019) 31043–31049. doi: 10.1021/acs.jpcc.9b08827
J. Long, S. Chen, Y. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 9711–9718. doi: 10.1002/anie.202002337
Q. Wu, H. Wang, S. Shen, et al., J. Mater. Chem. A 9 (2021) 5434–5441. doi: 10.1039/D0TA11209G
J.C. Liu, X.L. Ma, Y. Li, et al., Nat. Commun. 9 (2018) 1610. doi: 10.1038/s41467-018-03795-8
C. Wei, Y. Sun, G.G. Scherer, et al., J. Am. Chem. Soc. 142 (2020) 7765–7775. doi: 10.1021/jacs.9b12005

Figure 1 (a) Structure of TM-N4 and the considered TM atoms. (b-d) Free energy diagrams of NO reduction to NH3 on V-N4, Fe-N4 and Cu-N4 under 0 V vs. RHE.

Figure 2 NORR over Fe-N4. (a) Geometries of the IS, TS and FS for NO* with H* to NOH* or HNO*. (b) Differential charge density plot and Bader charge analysis of NO adsorbed. (c) Free energy diagrams of NORR via HNO* or NHO*. (d) Projected density of states (PDOS) of Fe-3d orbitals for Fe-N4, Fe-3d and NO-2p orbitals for NO adsorbed on Fe-N4 and NO-2p orbitals for free NO molecule, respectively.

Figure 3 (a) Formation energy barrier of the decoration of Fe atom on different sites of N-doped graphene. (b) Variation of temperature and total energy of single Fe-N4 during AIMD simulation. The simulation was running under 300 K for 10 ps with a time step of 2 fs.

Figure 4 (a) Thermodynamic estimation and screening catalysts of NO to NH3 via ∆G plotted versus Gads(N*). (b) A two-dimensional activity map for ammonia production.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
