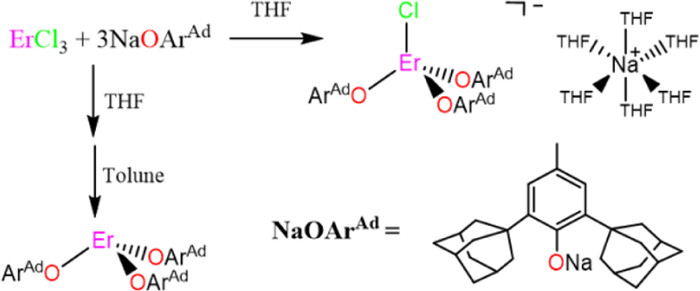
Scheme 1.
Synthesis of complexes 1 and 2.
Switching the coordination geometry to enhance erbium(Ⅲ) single-molecule magnets
Qian-Cheng Luo , Ning Ge , Yuan-Qi Zhai , Tengbo Wang , Lin Sun , Qi Sun , Fanni Li , Zhendong Fu , Yan-Zhen Zheng

Single-molecule magnets (SMMs), a type of magnetic nano-materials self-assembled by coordination chemistry, is appealing due to their inherent characteristics of possessing magnetic bistability and slow magnetic relaxation processes. The feature of storing bytes information relaying on single molecule improves information storage density, making it possible for the applications of spintronic devices and quantum information processing (QIP) [1-3]. Since the mononuclear complex [TbPc2]− (Pc = dianion of phthalocyanine), whose effective energy barrier (Ueff) is 230 cm−1, was reported by Ishikawa et al. [4], lanthanide cations, such as Tb(Ⅲ), Dy(Ⅲ), Ho(Ⅲ) and Er(Ⅲ), have been attached importance to construct high performance SMMs in the light of their high magnetic anisotropy and strong spin-orbit coupling effects [5-9]. For almost a decade, enormous progress for enhancing Ueff and magnetic blocking temperature (TB) has been made for lanthanide-based complexes, which are almost Dy(Ⅲ) compounds [10,11]. Especially lately, a record-breaking mixed-valence dilanthanide complex (CpiPr5)2Dy2I3 with Ueff of 1631 cm−1 and TB of 72 K was reported by Gould et al. [12].
Despite the fact that the performance of Er(Ⅲ) SMMs is relatively poor by contrast with Dy(Ⅲ)-based counterparts, it is better than SMMs with other prolate Ln(Ⅲ) ions. A series of reported mononuclear Er(Ⅲ) SMMs are listed in Tables S12 and S13 (Supporting information), where the highest Ueff of 300 cm−1 can be observed in [(C5H5BMe)Er(COT)] (COT = cyclooctatetraenyl) [13], and the maximum TB does not exceed 10 K in several (cyclooctatetraenyl) erbium(Ⅲ) complexes [14-16]. It is evident that the COT ligand can stabilize magnetic ground state of Er(Ⅲ) and makes those complexes with sandwich structure exhibit excellent SMM properties. Apart from those organometallic Er(Ⅲ) compounds, SMMs with monodentate coordination have also been exploited and reported all the time (Table S13). Zhang et al. prepared an equatorially nitrogen-coordinated complex with C3v local geometry Er[N(SiMe3)2]3 which possesses an energy barrier of 85 cm−1 in the absence of a static field [17]. Similarly, two three-coordinate Er(Ⅲ) compounds with oxygen and carbon donors were reported by Zhang et al. [18], namely Er(dbpc)3 (dbpc = tris(2, 6-di-tert-butyl-p-cresolate)) and Er(btmsm)3 (btmsm = tris(bis(trimethylsilyl)methyl)), exhibiting lower Ueff values of 39 and 80 cm−1, because of the conjugation effects and weak lone pairs for the former and soft carbon donor effect for the latter, respectively.
Stark energy level splitting is sensitive to the coordination sphere and appropriate crystal field environment plays a critical role to construct high-performance SMMs: Long and his co-workers point out that equatorial coordination environments is more conducive for those prolate 4f ions, for instance, Yb(Ⅲ) and Er(Ⅲ), because it can reduce and avoid charge contact between axially located electron density of Ln(Ⅲ) ions and coordination atoms [19]. Furthermore, the existence of quantum tunneling of the magnetization (QTM) leads to under barrier relaxation pathways and poor SMM properties, and high local symmetry around Ln(Ⅲ) ions is capable of suppressing such fast relaxation process which has been confirmed by both theoretical and experimental results [20-27]. Herein, considering prolate electron density of Er(Ⅲ), we utilized one variety of "hard" base ligand with large steric hindrance (-OArAd) and successfully isolated two complexes 1 ([ErCl(OArAd)3][Na(THF)6]) and 2 (Er(OArAd)3). Remarkably, ac susceptibility measurements reveal that out-of-phase (χ'') signals can be observed in complex 1 at dc magnetic field of 800 Oe and 2 at zero dc field, and field-induced Orbach barrier height of 2 arrives at 43(1) cm−1 (61(2) K).
The sterically demanding aryloxide ligand, AdArOH (AdArO = O-C6H2-2, 6-Ad-4-Me), was synthesized following a procedure from Watanabe et al. [28]. The corresponding deprotonated salt, NaOArAd, was prepared in 94% yield by addition of NaH to the phenol in THF suspension. The mixed aryloxide-chloride [ErCl(OArAd)3][Na(THF)6] (1) was prepared by metathesis of anhydrous ErCl3 with 3 equiv. of NaOArAd in THF, giving a pink crystal in 60% yield (Scheme 1). Treating 1 with toluene resulted in the formation of chloride free Er(OArAd)3 (2), which could be isolated as a pinkish crystal in 42% (Scheme 1).
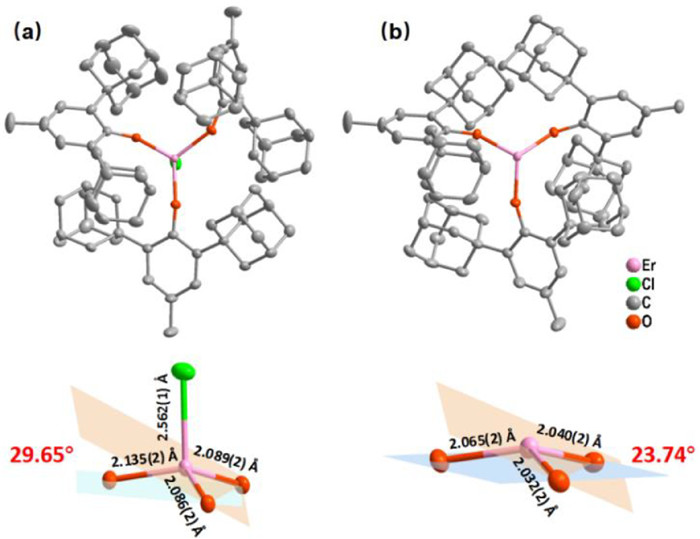
X-Ray single-crystal diffraction analysis reveals that 1 and 2 crystallize in the monoclinic P21/c and triclinic P1 space groups (Table S1), respectively. Compound 1 is composed of four-coordinate Er(Ⅲ) anions with distorted tetrahedral configuration ([ErCl(OArAd)3]−) and charge-balancing cations [Na(THF)6]+, while electrically neutral complex 2 is characteristic of a flat trigonal pyramid, in which Er(Ⅲ) ion is coordinated with three -OArAd ligands with large steric hindrance (Fig. 1). The Er-Cl bond length in 1 is 2.562(1) Å and the average distance of Er-O bonds is 2.103 Å. In contrast to 1, the elimination of Cl− ion leads to shorter average distance of Er-O bonds (2.046 Å) and smaller structural distortion relative to planar triangle geometry in 2 (Fig. 1 and Table S2 in Supporting information). These Er-O distances are close to several reported Er(Ⅲ) SMMs containing merely monodentate oxygen donors but significantly shorter than [L2Er(H2O)5][I]3·L2·(H2O) (L = tBuPO(NHiPr)2) and [Er(depma)2(H2O)6]Cl3·6H2O (depma = 9-diethylphosphono-methylanthracene) (Table S13). Meanwhile, three O-Er-O angles in both complexes range from 105.84(9)° to 120.27(9)° for 1 and 111.99(9)° to 119.93(9)° for 2. In a packing diagrams, the shortest intermolecular Er···Er distances are 12.954(1) and 14.747(1) Å (Figs. S1 and S2 in Supporting information), suggesting that both complexes are well isolated and intermolecular magnetic interactions are negligible.
The temperature dependent dc magnetic susceptibility measurements were carried out towards the powder samples of 1 and 2 at the temperature range of 2–300 K under an applied field of 1000 Oe (Figs. S3 and S4 in Supporting information). At the room temperature, the experimental χMT products of both compounds are 11.06 and 11.36 cm3 K/mol, respectively, which are agreement with theoretical value of 11.48 cm3 K/mol for free Er(Ⅲ) ion (S = 3/2, L = 6, g = 6/5). The χMT value undergoes a gradual decrease when lowering the temperature and arrives at the minimum of 9.75 and 8.69 cm3 K/mol at 2 K for 1 and 2 respectively, which can be attributed to the thermal depopulation of the Stark levels from Er(Ⅲ) ions [29]. A relatively more drastic drop of 2 than 1 at low temperature below 10 K can also be observed, indicating its characteristic of slower magnetic relaxation. The field dependent dc magnetization data of them were also measured from 0 to 5 T at 2 K (Figs. S5 and S6 in Supporting information). The magnetization increases linearly at low magnetic fields until around 0.5 T and reaches similar saturation values of 4.78 and 4.65 μB for 1 and 2, respectively.
Dynamic magnetic properties of 1 and 2 were investigated in the frequency range of 1–1218 Hz through ac magnetic susceptibility measurements. Under zero dc field, significant out-of-phase (χ'') signal can be observed only in complex 2 and the peak of χ'' shifts from 2 K to 10.19 K (Figs. S12 and S13 in Supporting information), demonstrating its slow magnetic relaxation behavior and SMM characteristic while strong QTM exists in 1 (Fig. S7 in Supporting information) [30]. To confirm the optimum field and study their dynamic magnetic behavior under dc field, ac measurements were performed under the magnetic field range of 0–2000 Oe at 2 K (for 1) and 5 K (for 2). Finally, the field of 800 and 1000 Oe were adopted for 1 and 2 in the light of measured strongest out-of-phase signals (Figs. S8 and S14 in Supporting information). Under this condition, frequency dependence of χ'' peaks can be observed from 3.57 K to 8.25 K for 1 and 4.92 K to 12.50 K for 2 in measured frequency range (Figs. S10 and S16 in Supporting information). Then relaxation times (τ) were extracted by the generalized Debye model and all Cole-Cole plots indicates the presence of one relaxation process (Figs. S11, S17 and S18 in Supporting information). Further investigation of magnetic relaxation processes can be achieved by the temperature dependence of τ and analysed by the plots of τ−1 vs. T (Figs. S19–S22 in Supporting information). For 1 under dc field of 800 Oe, its relaxation behavior can be well-modelled only considering individual Raman process (Eq. 1). An additional term to account for QTM process was required to fit the relaxation behavior of 2 under zero dc field due to slow variation of τ−1 value in the low temperature region (Eq. 2). Moreover, for 2 under dc field of 1000 Oe, only the combination of Orbach and Raman mechanisms can well describe the relationship between τ−1 and T (Eq. 3). The best fit parameters are summarized in Table 1. It is obvious that the coordination of Cl− ion induces strong QTM process and the elimination of this coordinated chloride suppresses the fast relaxation processes, leading to field-induced effective energy barrier value (Ueff) of 2 arrives at 43(1) cm−1 (61(2) K).
|
|
(1) |
|
|
(2) |
|
|
(3) |
Complete-active-space self-consistent filed spin-orbit (CASSCF-SO) calculations were performed using OPEN MOLCAS package [31] to further understand magnetic relaxation mechanisms of 1 and 2. Evidently, calculated g-tensors of the ground Kramers' doublet (KD) for both complexes are gx = 0.03, gy = 0.04, gz = 17.82 for 1 and gx = gy = 0.02, gz = 17.80 for 2 (Tables S3 and S4 in Supporting information), indicating they possess large magnetic anisotropy. Meanwhile, the predominant wavefunction composition (89.5% |±15/2 > for 1 and 98.5% |±15/2 > for 2) and the direction of principal magnetic axes (Figs. S23 and S24 in Supporting information) for the ground KD also verify this. We find that the orientation of easy axis is not exactly along Er-Cl bond for the former or perpendicular to the plane composed of three coordinated O atoms for the latter, existing a certain deviation, which could be caused by the structural distortions compared with the strict local symmetry of T or C3v from the perspective of molecular geometric configuration [32]. For 1, the admixture of wavefunction emerges at the first excited KD (63.8% |±13/2 > + 18.7% |±11/2 > ) and relatively significant transition magnetic moment between this KD arrives at ~10−1 μB, therefore a fast QTM process will occur at this KD theoretically, making the calculated Ueff is around 61 cm−1 (88 K) (Table S9 and Fig. S27a in Supporting information). Nevertheless, non-ignorable transition probability of 1.20 × 10−2 μB between the ground KD can be also observed, hinting that a non-vanishing QTM may exist. In reality, two-phonon Raman process also participate in the magnetic relaxation in the temperature range of ac measurement at the same time. Computed LoProp charge of donor Cl using the CASSCF wavefunctions is up to −0.8303, and the poor SMM property of 1 can be attributed to the energy destabilization of the prolate Er(Ⅲ) ion induced by rather strong repulsion with axial highly charged Cl− ion (Fig. S26 in Supporting information) [19,33]. Accordingly, with axial ligand eliminated, the coordination environment of 2 containing merely three O atoms is more suitable for Er(Ⅲ) ion: The energy gap between the ground KD and the first low-lying state is greatly improved to 155 cm−1 (223 K) (Table S4), and the absolute value of axial crystal-field parameters (CFPs) of 2 (B20 = −3.32 cm−1, B40 = 2.37 × 10−3 cm−1 and B60 = −2.81 × 10−5 cm−1) are significantly larger than 1 (B20 = −1.94 cm−1, B40 = 1.27 × 10−3 cm−1 and B60 = −6.78 × 10−6 cm−1) and smaller magnitude of non-axial CFP (Bkq, q ≠ 0) can be observed in 2 (Tables S6 and S7), indicating stronger axial anisotropy in 2. Besides, the admixture of wavefunction in 2 does not emerge until the fourth excited KD (78.5% |±7/2 > ), and the principal magnetization axis of this doublet is also largely tilted from the ground KD. Despite this, transition probability of 1.07 × 10−1 μB between the second excited KD indicates the occurrence of QTM process in this KD and computed energy barrier of 224 cm−1 (322 K) (Table S10 and Fig. S27b in Supporting information). Similar to complex 1, although calculated Ueff value is larger than the experimental value, it is indeed qualitatively reasonable in terms of the order if not considering other existing effects, such as off-resonance phonon modes and hyperfine interactions [34,35].
Then, their relaxation times for QTM process at the ground KD were evaluated theoretically through Eqs. 4 and 5 proposed by Yin et al. [36]. Here, β is the Bohr magneton, h is Planck constant and the gx, gy and gz are components of g tensor for the ground KD in x, y and z directions. The magnitude of the magnetic fields induced by dipolar and hyperfine interactions (Have) is estimated to be 20 mT because ab initio calculations can not provide this parameter from x to z directions directly and this selected value which can predict τQTM well was utilized in previously several reported systems [37,38]. The calculated τQTM of 1 is 4.63 × 10−5 s while this relaxation time for 2 (2.29 × 10−4 s) is almost five times than 1, indicating faster QTM process occurs between the ground KD in 1 and it can explain why its slow magnetic relaxation behavior lacks under zero dc field. And computed τQTM for 2 is slightly smaller than the experimentally observed value at 2 K (8.57 × 10−4 s).
|
|
(4) |
|
|
(5) |
In spite of enhanced SMM performance in 2 realized by switching the coordination geometry from 1, it is still poorer than its analogues, namely Er[N(SiMe3)2]3, Er(btmsm)3 and Er(dbpc)3 (Table S13 in Supporting information). In our case, "hard" oxygen donor was utilized and the shifting is the lowest between Er(Ⅲ) and the ligand coordination plane, however, we can observe that strong QTM process at absence of external field and lower field-induced Orbach energy barrier under dc magnetic field. For the former, the local symmetry around Er(Ⅲ) ion in 2 is deviate more from C3v than above-mentioned complexes in terms of bond lengths and angles which exacerbates its transverse magnetic anisotropy and fast relaxation under low temperature (Table S2). Additionally, similar to Er(dbpc)3, the electron density of O donors with contracted lone pairs is delocalized on the aromatic ring, leading to the decrease of effective charge and lower Ueff value [18]. By contrast, as Yamashita et al. analyzed using the localized orbital locator (LOL), the lone pairs on coordinated C atoms in Er(btmsm)3 diffuse towards central Er(Ⅲ) and increase the effective charge, making it possess comparable Ueff value to Er[N(SiMe3)2]3. Under such circumstances, further enhancement of magnetic relaxation properties can be achieved by improving local symmetry when using aryloxide ligands.
To further weaken transverse anisotropy of low-lying doublets and investigate the influence of local symmetry around Er(Ⅲ) ion, model complex (3) with D3h local symmetry was constructed in the light of the structure of 2 (Fig. S27c in Supporting information) and calculation at the same level was also carried out. The Er-O bonds in 3 were fixed as the average value of those in 2 (2.046 Å) and each angle of O-Er-O is perfectly 120°. As expected, eight KDs span an energy barrier of 687 cm−1 (988 K) (Table S5 in Supporting information) and CFPs of complex 3 calculated by the SINGLE_ANISO program demonstrate its improved axiality with axial CFPs of B20 = −3.76 cm−1 and B40 = 3.29 × 10−3 cm−1, despite nearly consistent parameter B60 = −2.38 × 10−5 cm−1 with 2 (Table S8 in Supporting information). The calculated τQTM is up to 2.80 × 10−2 s and transition probability of 4.98 × 10−4 μB between the ground KD is small enough to be neglected (Table S11 in Supporting information). Above results demonstrate that improved local symmetry enlarges crystal field splitting of Er(Ⅲ) ion and effectively suppresses QTM process under zero field condition. Significant QTM can be observed at the fourth excited KD (7.77 × 10−1 μB) and leads to theoretical Ueff value arrives at 416 cm−1 (598 K) (Fig. S27d in Supporting information), almost twice as much as 2. This indicates that the crystal field environment with high local symmetry constructed by only coplanar equatorial coordinated atoms is conducive to greatly improve effective energy barrier of prolate Er(Ⅲ) SMMs and it can be realized through modifying ligands by chemical means.
To summarize, we successfully isolated two Er(Ⅲ) complexes by utilizing substituted phenols with large steric hindrance (-OArAd) and they possess different geometry configurations in THF and toluene, respectively. Magnetic measurements reveal SMM behavior of both compounds and field-induced Orbach barrier height of complex 2 is 43(1) cm−1 (61(2) K). Ab initio calculations towards model complex 3 suggest that the removal of axial electrostatic repulsion and coplanar equatorial coordination pattern with high local symmetry are beneficial to further suppress QTM process and increase the Ueff value. Future work would include constructing more ideal crystal field environment to meet Er(Ⅲ) ion with prolate electron density and improve the slow relaxation properties of complexes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by National Natural Science Foundation of China (Nos. 21971203, 82073271 and 81803026), Key Scientific and Technological Innovation Team of Shaanxi Province (No. 2020TD-001) and the Fundamental Research Funds for Central Universities.
Supplementary material associated with this article can be found, in the online version, at doi:
L. Bogani, W. Wernsdorfer, Nat. Mater. 7 (2008) 179–186. doi: 10.1038/nmat2133
M.R. Wasielewski, M.D.E. Forbes, N.L. Frank, et al., Nat. Rev. Chem. 4 (2020) 490–504. doi: 10.1038/s41570-020-0200-5
S.L. Bayliss, D.W. Laorenza, P.J. Mintun, et al., Science 370 (2020) 1309–1312. doi: 10.1126/science.abb9352
N. Ishikawa, M. Sugita, T. Ishikawa, S.Y. Koshihara, Y. Kaizu, J. Am. Chem. Soc. 125 (2003) 8694–8695. doi: 10.1021/ja029629n
V.S. Parmar, D.P. Mills, R.E.P. Winpenny, Chem. Eur. J. 27 (2021) 7625–7645. doi: 10.1002/chem.202100085
X. Liu, X. Feng, K.R. Meihaus, et al., Angew. Chem. Int. Ed. 59 (2020) 10610–10618. doi: 10.1002/anie.202002673
K. Liu, X.J. Zhang, X.X. Meng, et al., Chem. Soc. Rev. 45 (2016) 2423–2439. doi: 10.1039/C5CS00770D
X.X. Meng, M.M. Wang, X.S. Gou, et al., Inorg. Chem. Front. 8 (2021) 2349–2355. doi: 10.1039/d1qi00145k
S.W. Zhang, W. Shi, P. Cheng, Coord. Chem. Rev. 352 (2017) 108–150. doi: 10.1016/j.ccr.2017.08.022
F.S. Guo, B.M. Day, Y.C. Chen, et al., Science 362 (2018) 1400–1403. doi: 10.1126/science.aav0652
Y.S. Ding, N.F. Chilton, R.E.P. Winpenny, Y.Z. Zheng, Angew. Chem. Int. Ed. 55 (2016) 16071–16074. doi: 10.1002/anie.201609685
C.A. Gould, K.R. Mcclain, D. Reta, et al., Science 375 (2022) 198–202. doi: 10.1126/science.abl5470
Y.S. Meng, C.H. Wang, Y.Q. Zhang, et al., Inorg. Chem. Front. 3 (2016) 828–835. doi: 10.1039/C6QI00028B
L. Münzfeld, C. Schoo, S. Bestgen, et al., Nat. Commun. 10 (2019) 3135. doi: 10.1038/s41467-019-10976-6
L. Ungur, J.J. Le Roy, I. Korobkov, M. Murugesu, L.F. Chibotaru, Angew. Chem. Int. Ed. 53 (2014) 4413–4417. doi: 10.1002/anie.201310451
K.R. Meihaus, J.R. Long, J. Am. Chem. Soc. 135 (2013) 17952–17957. doi: 10.1021/ja4094814
P. Zhang, L. Zhang, C. Wang, et al., J. Am. Chem. Soc. 136 (2014) 4484–4487. doi: 10.1021/ja500793x
H.T. Zhang, R. Nakanishi, K. Katoh, et al., Dalton Trans. 47 (2018) 302–305. doi: 10.1039/C7DT04053A
J.D. Rinehart, J.R. Long, Chem. Sci. 2 (2011) 2078–2085. doi: 10.1039/c1sc00513h
S.K. Gupta, T. Rajeshkumar, G. Rajaraman, R. Murugavel, Chem. Sci. 7 (2016) 5181–5191. doi: 10.1039/C6SC00279J
M.A. Sørensen, U.B. Hansen, M. Perfetti, et al., Nat. Commun. 9 (2018) 1292. doi: 10.1038/s41467-018-03706-x
J.L. Liu, Y.C. Chen, Y.Z. Zheng, et al., Chem. Sci. 4 (2013) 3310–3316. doi: 10.1039/c3sc50843a
Z.H. Li, Y.Q. Zhai, W.P. Chen, Y.S. Ding, Y.Z. Zheng, Chem. Eur. J. 25 (2019) 16219–16224. doi: 10.1002/chem.201904325
X.L. Ding, Y.Q. Zhai, T. Han, et al., Chem. Eur. J. 27 (2021) 2623–2627. doi: 10.1002/chem.202003931
J. Wu, O. Cador, X.L. Li, et al., Inorg. Chem. 56 (2017) 11211–11219. doi: 10.1021/acs.inorgchem.7b01582
M.A. AlDamen, J.M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo, A. Gaita-Ariño, J. Am. Chem. Soc. 130 (2008) 8874–8875. doi: 10.1021/ja801659m
C.R. Ganivet, B. Ballesteros, G. de la Torre, et al., Chem. Eur. J. 19 (2013) 1457–1465. doi: 10.1002/chem.201202600
T. Watanabe, Y. Ishida, T. Matsuo, H. Kawaguchi, Dalton Trans. 39 (2010) 484–491. doi: 10.1039/B911082H
A.J. Brown, D. Pinkowicz, M.R. Saber, K.R. Dunbar, Angew. Chem. Int. Ed. 54 (2015) 5864–5868. doi: 10.1002/anie.201411190
J. Long, B.G. Shestakov, D. Liu, et al., Chem. Commun. 53 (2017) 4706–4709. doi: 10.1039/C7CC02213A
I.F. Galván, M. Vacher, A. Alavi, et al., J. Chem. Theory Comput. 15 (2019) 5925–5964. doi: 10.1021/acs.jctc.9b00532
S.K. Singh, B. Pandey, G. Velmurugan, G. Rajaraman, Dalton Trans. 46 (2017) 11913–11924. doi: 10.1039/C6DT03568J
T.A. Bazhenova, V.A. Kopotkov, D.V. Korchagin, et al., Molecules 26 (2021) 6908. doi: 10.3390/molecules26226908
A. Lunghi, F. Totti, R. Sessoli, S. Sanvito, Nat. Commun. 8 (2016) 14620.
F. Lu, M.M. Ding, J.X. Li, B.L. Wang, Y.Q. Zhang, Dalton Trans. 49 (2020) 14576–14583. doi: 10.1039/d0dt02868a
B. Yin, C.C. Li, Phys. Chem. Chem. Phys. 22 (2020) 9923–9933. doi: 10.1039/d0cp00933d
S. Zhang, J. Tang, J. Zhang, et al., Inorg. Chem. 60 (2021) 816–830. doi: 10.1021/acs.inorgchem.0c02863
Y. Dong, L. Zhu, B. Yin, X. Zhu, D. Li, Dalton Trans. 50 (2021) 17328–17337. doi: 10.1039/d1dt02925h

Figure 1 (Top) Crystal structures of (a) 1 and (b) 2. color codes: Er, pink; O, red; C, gray; Cl, green. All hydrogen atoms have been omitted for clarity. (Bottom) The diagrammatic sketch of ErO3Cl and ErO3 geometry for 1 and 2, respectively. The angles represent average dihedral angles between coordinate plane and center-donor plane.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载: