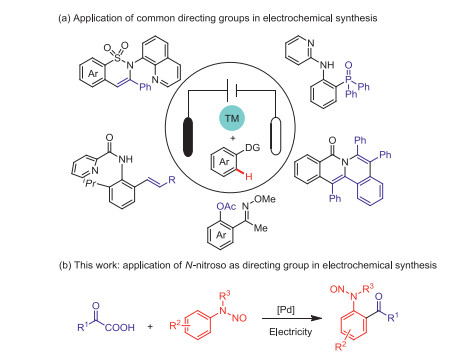
Figure 1.
Site-selective C–H activation via the use of a directing group of electrochemistry and transition-metal catalysis.
Electrochemically mediated decarboxylative acylation of N-nitrosoanilines with α-oxocarboxylic acids
Xinyu Wang , Shihong Wu , Yujing Zhong , Yingchun Wang , Yingming Pan , Haitao Tang

As the "holy grail" of organic synthesis, C–H functionalization has attracted wide attention from synthetic chemists [1]. Transition metal-catalyzed oxidative C–H functionalization provides a straightforward synthetic approach for the construction of carbon–carbon and carbon–heteroatom bonds [2, 3]. Especially, C–H functionalization of arenes can simplify the access to important biologically active small molecules [4, 5]. In recent years, direct functionalization of inert aryl C–H bonds under the catalysis of transition-metal catalysts has attracted extensive attention owing to their excellent step economy and atom efficiency [6-8], and aryl C–H activation methods with different directing groups have been successively developed [9]. Despite these achievements, many efficient directing groups have not yet been involved in C(sp2)–H functionalization. N-Nitroso compounds are a very useful class of medicinal compounds and synthetic materials [10, 11], and they are also important precursors for the synthesis of various nitrogen-containing compounds, such as hydrazine and octanone [12, 13]. Moreover, they are a potential directing group that can be applied to C–H activation [14, 15]. Therefore, the modification of N-nitroso compounds via C–H activation must be realized.
Electrochemistry has attracted increased attention in recent years because of its coincidence with the current development of green chemistry [16-21]. As a reliable alternative to redox reagents, electrochemistry can achieve redox conversions without exogenous oxidants, such as copper salts, silver salts, and persulfate salts [22-26]. In recent years, various electrooxidative transition metal catalytic systems that offer an efficient way to construct important chemical bonds have been developed (Fig. 1a) [27-31]. In these electrochemical reactions, pyridine, quinoline, oxime, and amide are used as C–H-activated directing groups. However, N-nitroso, a very active and plastic functional group, has not yet been studied by electrochemists. In this study, under electrochemical conditions, C–H acylation reaction was realized by taking it as a directing group. Hence, not only the traditional electrochemical synthesis of N-nitroso-2-aminobenzophenones under palladium catalysis and nonoxidation (Fig. 1b), but also flow electrochemical synthesis were achieved.
Phenylglyoxylic acid 1a and N-methyl-N-nitrosoaniline 2a were used as substrates. The reaction conditions were screened (Table 1). The specific information on the other conditions is provided in Table S1 (Supporting information). The results of electrolysis were optimal when nBu4NOAc (1.0 equiv.) was used with Pd(OAc)2 (10 mol%) as the catalyst, HFIP/TFE (3:3) as a solvent, Pt as an anode, and stainless steel as a cathode at a constant current of 5 mA and a reaction temperature of 60 ℃ in an undivided cell. The yield decreased to 63% when HFIP was used as a solvent (entry 2). Platinum plates and stainless steel or graphite rods were applied to both electrodes to test the electrode effect. The anode and the cathode were also changed, but none of the tested materials could increase the yield of 3aa (entries 3 and 4). When the temperature was changed to room temperature (entry 5), the yield of 3aa was 70%. Among the Pd catalysts tested, Pd(OAc)2 was found to be optimal (entries 6 and 7). When the amount of Pd(OAc)2 was reduced to 5 mol%, 3aa was afforded in 77% yield (entry 8). Control experiments showed that in the absence of electricity or Pd(OAc)2, no desired product was obtained (entries 9 and 10). The change in yield was also tested under divided cell conditions (entry 11). Considering whether the presence of acetate can promote the reaction, we tried to use HOAc as the additive. Unfortunately, the yield of the target product could not be improved (Table S1 in Supporting information).
Under the optimized reaction conditions, substrate suitability studies were conducted using α-oxocarboxylic acid compounds with various substituents (Scheme 1). First, an α-oxocarboxylic acid with a substituent in the ortho-, meta-, or para-position of the phenyl ring exhibited a good yield (3ab, 3aj and 3ak). Both electron-donating (OMe) and electron-withdrawing groups (F, Cl, Br, CN, COOMe and Ph) were also well tolerated, providing the corresponding products in 61%–88% yields (3ac–3ai). Furthermore, bearing a bis- or a tri-substituted phenyl unit was suitable for this reaction to afford 3al–3am in moderate yields. Moreover, 2-oxo-2-(thiophen-2-yl)acetic acid, 2-(furan-2-yl)-2-oxoacetic acid, and 2-(benzo[b]thiophen-2-yl)-2-oxoacetic acid smoothly participated in this reaction to give heteroaromatic ketones 3an, 3ao and 3ap in yields of 74%, 50% and 52%, respectively. Pyruvic acid could also be involved in the reaction and afforded the desired product in 51% yield (3aq). Additionally, we also tried 2-oxo-2-(pyridin-4-yl) acetic acid and other types of carboxylic acids (benzoic acid, phenylaceticacid, trans-cinnamic acid), but failed to get the target product (Scheme S1 in Supporting information).
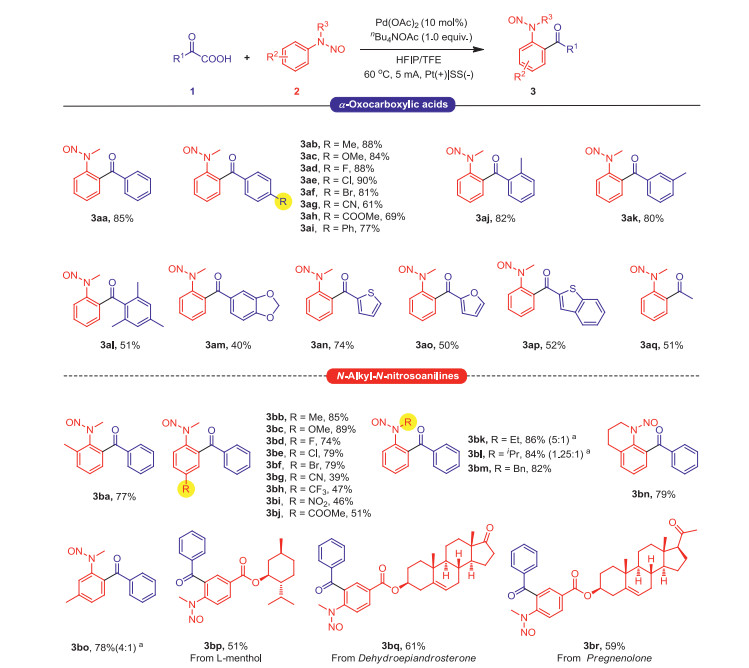
Subsequently, a substrate scope study of N-alkyl-N-nitrosoanilines compounds was conducted. As shown in Scheme 1, various N-alkyl-N-nitrosoaniline compounds afforded moderate to good yields under the electrochemical conditions. According to all the presented examples, the electronic properties of the substituents on the N-alkyl-N-nitrosoanilines slightly affected the yield of the product. For various N-alkyl-N-nitrosoanilines, the Me (3ba-3bb), MeO (3bc), F (3bd), Cl (3be), Br (3bf), CN (3bg), CF3 (3bh), NO2 (3bi), and COOMe (3bj) substituted N-alkyl-N-nitrosoanilines were suitable for this conversion, a result that also demonstrated the potential of this electroorganic synthesis method in organic synthesis. Different alkyl-substituted N-nitrosoanilines were also reacted with α-oxocarboxylic acids to obtain the target products in good yields (3bk–3bm). When the nitrogen atom has an ethyl or isopropyl group, the steric hindrance of the two groups on the nitrogen atom may be equal, resulting in cis and trans isomers (3bk-3bl) [15]. The N-nitrosoaniline corresponding to tetrahydroquinoline, the target product, could also be obtained in 79% yield (3bn). In addition to the ortho and para alkane groups, when alkane groups were in the meta-position, good selectivity could be obtained, that is, greater than 4:1 (3bo: 3bo').
Afterward, the substrate scope of natural product derivatives was investigated. The three natural product derivatives L-menthol, dehydroepiandrosterone, and pregnenolone gave the target product in moderate yields (3bp–3br), another result that demonstrated the potential of this electroorganic synthesis method in functional group tolerance.
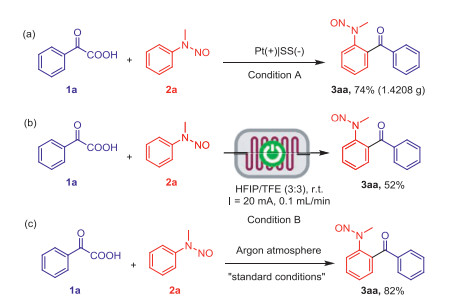
The utility of this reaction (Scheme 2) was further investigated by performing gram-scale-up experiments. The target product was obtained in moderate yields. Under standard conditions in the gram-scale experiments, the target product was obtained in good yield. However, the limitations of traditional electrochemistry, such as large electrode gap, limited mass transfer, and difficulty in scaling up, limit the industrial application of these complex organic chemical reactions. Electrochemical microreactors can satisfactorily solve these problems because of their extremely large surface-to-volume ratio, which enhances mass and heat transfer and provides more reaction sites compared with classical batch-type reactors [32]. Thus, this reaction was tested using a flow electrochemical reactor. Surprisingly, the target product was obtained in 52% yield. The reaction mechanism was further clarified by performing a series of control experiments (Scheme 2c). Additionally, 82% yield was obtained when the reaction was performed under Ar conditions. Therefore, this process was confirmed to be an electrooxidation process.
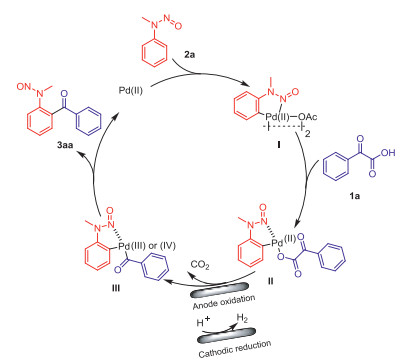
On the basis of the abovementioned results and the reports in the literature [15], a possible mechanism for the electrochemical oxidation is presented in Scheme 3. First, this transformation is believed to start with the ortho-palladation of 2a with Pd(OAc)2 to provide the five-membered palladacycle I, which subsequently undergoes anion exchange with 1a to afford intermediate II, followed by electrooxidation and concurrent decarboxylation to furnish the Pd(Ⅲ) or Pd(Ⅳ) intermediate III. Finally, product 3aa is generated by reductive elimination with the simultaneous release of a Pd(Ⅱ) species to complete the catalytic cycle. At the same time, protons were reduced at the cathode to produce H2.
In summary, we developed an economical and practical method for the synthesis of N-nitroso-2-aminobenzophenones through electrochemical oxidation. A wide range of N-nitroso-2-aminobenzophenones were obtained in moderate to high yields. The three natural product derivatives L-menthol, dehydroepiandrosterone, and pregnenolone gave the target product in moderate yields. We contend that this electrochemical strategy for accessing N-nitroso-2-amino benzophenones will facilitate the synthesis of various natural product compounds.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the Guangxi Natural Science Foundation of China (No. 2021GXNSFFA220005), the Central Government Guides Local Science and Technology Development Fund Projects (No. guike ZY21195014), National Natural Science Foundation of China (Nos. 22061003, 22161008, 22161007), middle-aged and young teachers' basic scientific research ability improvement project of Guangxi (No. RZ1900005748), the Opening Project of Hunan Engineering Laboratory for analyse and Drugs Development of Ethnomedicine in Wuling Mountain (No. hgxy2101) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Qin, L. Zhu, S. Luo, Chem. Rev. 117 (2017) 9433–9520. doi: 10.1021/acs.chemrev.6b00657
K.M. Engle, T.S. Mei, M. Wasa, J.Q. Yu, Acc. Chem. Res. 45 (2012) 788–802. doi: 10.1021/ar200185g
L. McMurray, F.O. Hara, M.J. Gaunt, Chem. Soc. Rev. 40 (2011) 1885–1898. doi: 10.1039/c1cs15013h
H. Schönherr, T. Cernak, Angew. Chem. Int. Ed. 52 (2013) 12256–12267. doi: 10.1002/anie.201303207
C.S. Leung, S.S.F. Leung, J. Tirado-Rives, W.L. Jorgensen, J. Med. Chem. 55 (2012) 4489–4500. doi: 10.1021/jm3003697
T. Gensch, M.N. Hopkinson, F. Glorius, J. Wencel-Delord, Chem. Soc. Rev. 45 (2016) 2900–2936. doi: 10.1039/C6CS00075D
F. Wang, S. Yu, X. Li, Chem. Soc. Rev. 45 (2016) 6462–6477. doi: 10.1039/C6CS00371K
Y. Wei, P. Hu, M. Zhang, W. Su, Chem. Rev. 117 (2017) 8864–8907. doi: 10.1021/acs.chemrev.6b00516
J.R. Hummel, J.A. Boerth, J.A. Ellman, Chem. Rev. 117 (2017) 9163–9227. doi: 10.1021/acs.chemrev.6b00661
S. Moncada, R.M. Palmer, E.A. Higgs, Pharmacol. Rev. 43 (1991) 109.
Z. Guo, M. Xian, W. Zhang, A. McGill, P.G. Wang, Bioorg. Med. Chem. 9 (2001) 99–106. doi: 10.1016/S0968-0896(00)00222-4
W.W. Hartman, L.J. Roll, Org. Synth. 13 (1933) 82. doi: 10.15227/orgsyn.013.0082
H. Wagner, J.B. Hill, J. Med. Chem. 17 (1974) 1337–1338. doi: 10.1021/jm00258a023
Y. Wu, L. Sun, Y. Chen, et al., J. Org. Chem. 81 (2016) 1244–1250. doi: 10.1021/acs.joc.5b02535
J.P. Yao, G.W. Wang, Tetrahedron Lett. 57 (2016) 1687–1690. doi: 10.1016/j.tetlet.2016.03.009
C. Kingston, M.D. Palkowitz, Y. Takahira, et al., Acc. Chem. Res. 53 (2020) 72–83. doi: 10.1021/acs.accounts.9b00539
J. Lia, S. Zhang, K. Xu, Chin. Chem. Lett. 32 (2021) 2729–2735. doi: 10.1016/j.cclet.2021.03.027
Y. Adeli, K. Huang, Y. Liang, et al., ACS Catal. 9 (2019) 2063–2067. doi: 10.1021/acscatal.8b04351
N. Chen, H.C. Xu, et al., Green Synth. Catal. 2 (2021) 165–178. doi: 10.1016/j.gresc.2021.03.002
Y. Wu, J.Y. Chen, H.R. Liao, et al., Green Synth. Catal. 2 (2021) 233–236. doi: 10.1016/j.gresc.2021.03.006
Z. Yang, Y. Yu, L. Lai, et al., Green Synth. Catal. 2 (2021) 19–26. doi: 10.1016/j.gresc.2021.01.009
Z.L. Wu, J.Y. Chen, X.Z. Tian, et al., Chin. Chem. Lett. 33 (2022) 1501–1504. doi: 10.1016/j.cclet.2021.08.071
Y. Yu, Y. Jiang, S. Wu, et al., Chin. Chem. Lett. 33 (2022) 2009–2014. doi: 10.1016/j.cclet.2021.10.016
Z. Li, Q. Sun, P. Qian, et al., Chin. Chem. Lett. 31 (2020) 1855–1858. doi: 10.1016/j.cclet.2020.02.030
J. Jiang, Z. Wang, W.M. He, Chin. Chem. Lett. 32 (2021) 1591–1592. doi: 10.1016/j.cclet.2021.02.067
M. He, P. Zhong, H. Liu, et al., Green Synth. Catal. (2022), doi: 10.1016/j.gresc.2022.03.002.
J. Frey, X. Hou, L. Ackermann, Chem. Sci. 13 (2022) 2729–2734. doi: 10.1039/D1SC06135F
Z.J. Wu, F. Su, W. Lin, et al., Angew. Chem. Int. Ed. 58 (2019) 16770–16774. doi: 10.1002/anie.201909951
K.J. Jiao, Y.K. Xing, Q.L. Yang, H. Qiu, T.S. Mei, Acc. Chem. Res. 53 (2020) 300–310. doi: 10.1021/acs.accounts.9b00603
Y. Cao, Y. Yuan, Y. Lin, et al., Green Chem. 22 (2020) 1548–1552. doi: 10.1039/D0GC00289E
Z.Q. Wang, C. Hou, Y.F. Zhong, et al., Org. Lett. 21 (2019) 9841–9845. doi: 10.1021/acs.orglett.9b03682
M. Elsherbini, T. Wirth, Acc. Chem. Res. 52 (2019) 3287–3296. doi: 10.1021/acs.accounts.9b00497

Figure 1 Site-selective C–H activation via the use of a directing group of electrochemistry and transition-metal catalysis.

Scheme 1 Substrate scope. Reaction conditions: Pt plate anode (1 cm × 1 cm), stainless steel cathode (1 cm × 1 cm), undivided cell, 1 (0.3 mmol), 2 (0.2 mmol), HFIP (3 mL), TFE (3 mL), nBu4NOAc (0.2 mmol), Pd(OAc)2 (0.02 mmol), 5.0 mA, 60 ℃, 3.5 h. Isolated yield. a Ratio of isomers, determined by the 1H NMR spectra.

Scheme 2 Applied and control experiments. Conditions A: Pt plate anode, stainless steel cathode, undivided cell, 1a (12 mmol, 1.8016 g), 2a (8 mmol, 1.0892 g), HFIP (45 mL), TFE (45 mL), nBu4NOAc (6 mmol), Pd(OAc)2 (0.8 mmol), 40 mA, 60 ℃, 20 h. Isolated yield. Conditions B (Electrolysis conditions): Graphite anode, stainless steel cathode, electrode surface (8 cm × 6 cm), 1a (0.3 mmol), 2a (0.2 mmol), solvent (6 mL), tr = 75 s (calculated), 60 min (3.7 F/mol). Isolated yield.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
