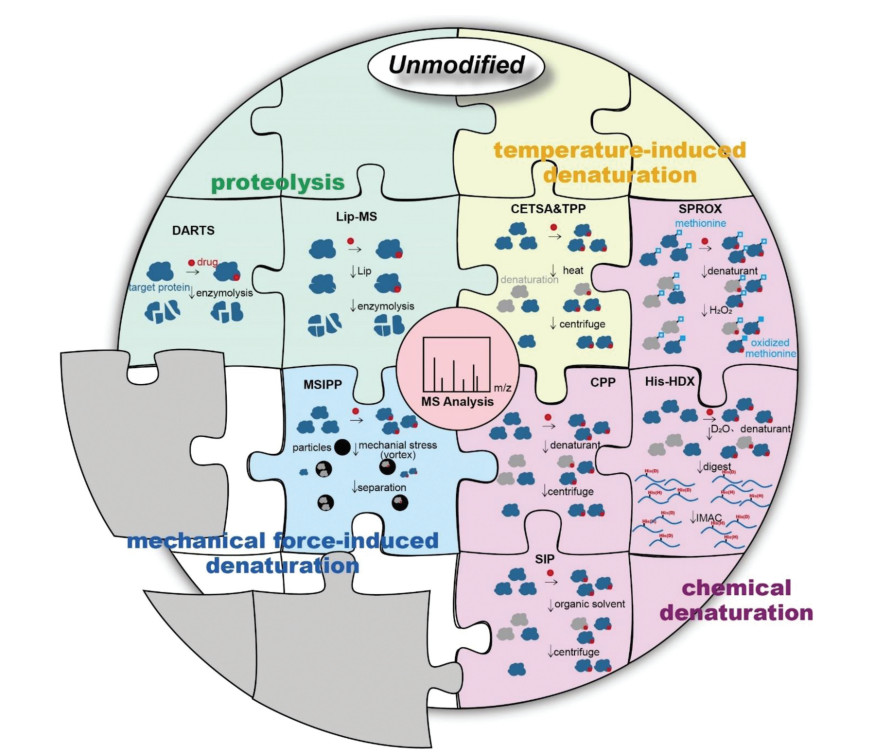
Figure 1.
An illustration of main unmodified methodologies for identifying and validating drug targets. The gray sections represent new methods to be developed.

Small-molecule drugs are organic compounds with the molecular weight below 900 daltons. They affect biological processes and constitute the vast majority of current drugs [1]. However, numerous targets of small-molecule drugs are still waiting to be discovered [2]. Therefore, the discovery and confirmation of protein targets are of great significance for drug discovery, structural optimization, and the disclosure of related pathways.
Current drug discovery can be classified into two categories, i.e., phenotypic drug discovery (PDD) and target-based drug discovery (TDD) [3]. The former explores the target and mechanism of the drug relying on the change of biological phenotypes upon the administration of the drug. Many currently used drugs are discovered by this strategy [4], such as artemisinin. Nevertheless, the discovery of the target and mechanism based on PDD still remains elusive, because the relationship between target-drug interaction and the resulting biological phenotype is still in a "black box". Differently from PDD, TDD obtains active compounds based on the spatial structure of the target protein, and relies more on the methods of molecular docking simulation, chemical synthesis, and machine learning [5-7]. Although TDD provides a relatively straightforward method for drug screening, there are still many unrevealed target proteins in the body as many drugs exert pharmacological effects by binding to multiple targets [8, 9]. Those unrevealed targets have been proved one of the important reasons for off-target effect and adverse reaction, which may lead to the limited efficacy and failure of treatment [4, 10]. Therefore, to develop more accurate and effective methods for the precise discovery and effective verification of targets are greatly important for the drug research and development.
In recent decades, great efforts have been made in method development for drug target discovery [11, 12]. Direct target identification methods can be mainly divided into two categories, namely modified and unmodified methods. The common modified method involves the covalent immobilization of drugs on a selected stationary phase (e.g., agarose, magnetic beads or microplates) and capture potential targets among matrices based on affinity. It includes affinity chromatography [13, 14], activity-based protein profiling (ABPP) [15-18], and protein microarray [19] methods, nanomaterials [20, 21] have also been used in affinity purification instead of agarose due to their unique properties [22-24]. However, the covalent immobilization of drugs in the modified method may change their chemical structures and interfere the interactions between drugs and targets, which requires efficacy validation before the target discovery study [25]. To address this limitation, a variety of target discovery methods using native chemical structure of drugs without modification have emerged. These methods include drug affinity responsive target stability (DARTS) [26], limited proteolysis-mass spectrometry (LiP-MS) [27], stability of proteins from rates of oxidation (SPROX) [28], cellular thermal shift assay (CETSA) [29] and so forth. Proteins undergo property changes after drug binding, such as conformational changes or sensitivity to proteolysis. Comparing the difference of proteomics before and after drug combination can screen out potential target proteins with characteristic changes. No need to derive the drug on the stationary phase, so these methods effectively reduce non-specific interactions and false positive results, showing significant advantages. Besides, they can simultaneously detect target proteins that directly interact with drug molecules and that are related to downstream pathways.
In this review, we will discuss these unmodified methodology advances for the target discovery of active molecules, mainly small molecule drugs, highlighting their achievements and limitations in drug development. According to the protein properties applied by various methods, we divide them into four categories, as shown in Fig. 1. Identifying targets at the initial stage of the drug development process establishes the link between drug activity and cell phenotype, and clarifies the drug action mechanism, which aid in the development of novel pharmacotherapy. Furthermore, screening 'unexpected' targets helps to predict potential toxicity and side effects. If necessary, drug structural adjustments can be made to reduce drug development costs. The in-depth elaboration and analysis of methodologies for the target discovery will provide valuable references for improving the efficacy and safety of drugs, and facilitate the development of potential drug therapies.
Under physiological conditions, the protein has multiple alternative conformations in a state of dynamic equilibrium and may also exhibit a certain degree of local reversible unfolding. When the recognition site of target proteins is masked by the drug of interest, the free energy of the protein-drug complex changes due to the forces such as hydrogen bond, hydrophobic and electrostatic interaction formed between them. The balance of binding and dissociation will drive the potential target protein to form a thermodynamically more stable conformation, which is very conducive to drug binding (Fig. 2A).
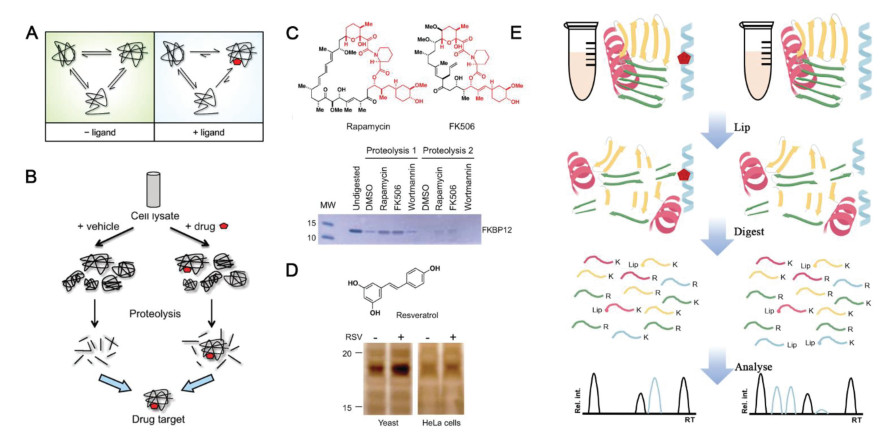
DARTS is a widely accepted method of drug target discovery. The potential target protein is stabilized in a different conformation from the drug-free state, which significantly enhances the resistance to enzymatic hydrolysis (Fig. 2B) [30]. The reduced protease sensitivity makes the potential target protein easy to be recognized by showing a condensed band on SDS-PAGE and subsequently identified by MS. DARTS was firstly proposed in 2009 by Huang and the co-workers [26, 31] and its feasibility was verified by the known target protein of rapamycin and FK506, FKBP12 (Fig. 2C). A new target of resveratrol, a translation factor eIF4A, was discovered by the DARTS method (Fig. 2D) [26], which revealed the protein translation mechanism of resveratrol for life-extending and provided theoretical basis for health care and disease prevention. Compared with other methods, the drug target discovery based on DARTS is easy to operate and it is compatible to biological systems. In addition, it is a universal method that does not require chemical labeling [32-34].
The discovery and mechanism research of obesity-associated protein (FTO) for the treatment of acute myelocytic leukemia (AML) is a typical example of DARTS application. (R)-2-Hydroxyglutarate (R-2HG), commonly considered as a cancer-promoting metabolite, was explored to competitively inhibit the activity of FTO, a demethylase, via directly acting on fat mass based on DARTS in 2017. It thereby increases the modification level of cellular N6-methyladenosine (m6A) RNA and results in the decrease of oncogene MYC. This result confirmed the "double-edged sword" effect of R-2HG on cancer and provided new ideas for the treatment of leukemia [35, 36]. The following year, this team and others developed two novel small molecule inhibitors of FTO, FB23 and FB23-2, which bound directly to FTO and specifically inhibit the m6A demethylase activity of FTO and inhibit the proliferation of AML (acute myelocytic leukemia) cells [37], which prompted to screen for more effective FTO inhibitors for clinical use. In 2020, two more small molecular compounds, bisantrene and brequinara were screened as FTO inhibitors and demonstrated by DARTS to bind directly to FTO. They have significant therapeutic effects on multiple AML mouse models, especially relapsed PDX (patient-derivedxenografts) models. In addition, LILRB4 was found to be the direct target gene of FTO, and the expression abundance of LILRB4 was 40–50 times higher than that of PD-L1/PD-L2, which partly explained why the immunotherapy strategy targeting PD-1/PD-L1/PD-L2 in previous clinical trials failed to achieve significant efficacy in AML [38]. This series of studies provided a new strategy for the clinical treatment of AML.
Additionally, dihydroorotate dehydrogenase (DHODH) was protected from protease degradation in a dose-dependent manner by the lead compound RYL-634 in a DARTS study. Since DHODH was verified as an antiviral target in the previous research, there was great potential to develop RYL-634-based antiviral drugs [39]. What is more, DARTS was used to identify lamin A/C, ATP-dependent RNA helicase DDX1 (DDX1), heterogeneous nuclear ribonuclear protein H1/H2 (hnRNP H2) and heterogeneous nuclear ribonuclear protein A2/B1 (hnRNP A2/B1) as targets of the compound 2155-14, thus providing a therapeutic approach for melanoma [40].
However, the current DARTS method still has some limitations: (1) Low-abundance target proteins would be missed due to the limited detection sensitivity; (2) Not suitable for the discovery of target proteins with weak affinity for drug binding; (3) The effectiveness of the method for target proteins that are extremely sensitive or resistant to the protease still needs to be verified; (4) It is only suitable for targets with significantly variational bands after incubation with the drug, otherwise limited sensitivity would result in false negative results [11, 26].
LiP-MS was developed as a high throughput strategy for drug target discovery and identification in biofluids [41]. In Lip-MS, broad-specificity proteases such as proteinase K and thermolysin are preliminarily applied for limited proteolysis. The resulted structure-specific fragments are subjected to secondary complete digestion by trypsin that selectively cleaves the C-termini of lysine and arginine [42]. Drug-induced protein structural alternation would lead to cleavage site difference during limited proteolysis [43]. Proteins that suffering proteolytic pattern variation or having detectable abundant difference of resulted peptides upon drug binding are considered as potential targets (Fig. 2E) [44]. In addition, the obtained enzymatic digestion sites were informative for drug binding site determination [27, 43]. Moreover, its application was not limited to drug target discovery, but also extended to the identification of metabolite-protein interactions. Through unbiased LiP technology, the Cdc19 protein (pyruvate kinase) was discovered as target of the endogenous fructose-1, 6-bisphosphate (FBP), which underwent structural transitions. Additionally, after the addition of FBP, the abundance of 23 peptides of fatty acid synthase subunit 1 (Fas1) changed more than 5 times. It was confirmed that FBP directly interacts with yeast FAS complex by LiP-MS to cause conformational changes of core proteins in metabolic pathway [42]. LiP-MS is simpler than DARTS which requires SDS-PAGE analysis and in-gel digestion, parameters including the ratio of two proteases, concentrations, and temperature control should be carefully considered, which may confuse the enzymatic sites, making it challenge to obtain reproducible results.
After heated, the proteins undergo denaturation and precipitation throught irreversibly lose the folded structures or expose the hydrophobic core. The thermodynamic stability of the protein changed after binding with the drug. Just as the drug-bound proteins in DARTS enhance their resistance to proteolysis, they also enhance the resistance to temperature-induced denaturation. Based on the principle, researchers developed the following methods.
The CETSA technology was first proposed in 2013 [29, 45], upon which the target protein discovery was achieved by thermal regulation. Heating aliquots of cell lysate to different temperatures, soluble proteins can be separated from the precipitated ones by centrifugation. After the target protein is relatively quantified by western blotting, a protein melt curve can be developed according to the treated temperature and relative band intensity. Proteins with significant shift of melting curves upon drug binding were suggested as potential targets [46]. This method was compatible with biological systems, such as intact cells or animal tissue [29]. Acetaminophen was verified to bind weakly with quinone reductase 2 (NQO2) by CETSA in living cells, thus NQO2 was proved to be the off-target of acetaminophen. As NQO2 is highly expressed in liver and kidney and regulates the level of reactive oxygen species in cells, which explains the acute hepatotoxicity caused by acetaminophen overdose [47]. In addition, CETSA had also been applied in vivo to evaluate the binding of the lead compound of receptor interacting protein 1 kinase (RIPK1) to RIPK1, thus monitoring the in vivo binding of drug targets in the spleen and brain of mice (Fig. 3A), providing an important basis for preclinical drug development [48]. Nonetheless, immunoblotting experiments have to rely on suitable antibodies and probably miss targets with low-abundance, which limits the commonality of this method. And CETSA is often used for target identification, but it still has the limitation of insufficient flux for discovering new targets.
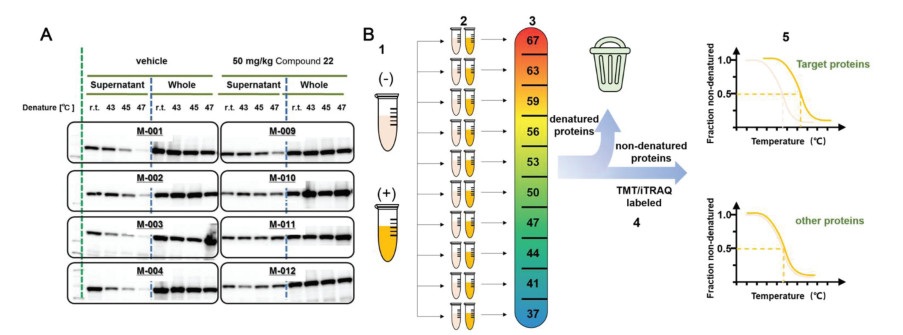
To overcome the shortcomings and challenges of CETSA, Savitski research group combined CETSA with quantitative MS to develop a high-throughput target screening method named thermal proteome profiling (TPP), also called CETSA-MS [49]. The exploration of TPP and quantification of separated proteins were achieved by involving quantitative proteomic labeling technologies, such as isobaric tags for relative and absolute quantitation (iTRAQ) and tandem mass tag labels (TMT). The quantity of soluble protein as a function of the temperature was then plotted to get the melting curve of proteins. The temperature corresponding to the denaturation of 50% soluble protein was recorded as the melting temperature (Tm). Thus, two melting curves were obtained with drug-bound and drug free states, and the calculated melting point difference (ΔTm) between them suggested the effective combination of the ligand and the target (Fig. 3B) [49,50]. However, the designed strategy was not suitable for water-insoluble membrane proteins, until the introduction of small amount of detergent. 0.4% Nonidet P-40 was applied for membrane protein extraction without causing chemical denaturation or reconstitution of aggregated proteins, which extended the application of this strategy to membrane proteins [51]. Later in 2022, precipitate-supported thermal proteome profiling (PSTPP), which was evolved from TPP, was proposed for comprehensive screening and identification of drug targets. In this method, the precipitated and soluble components produced in the experiment are both effectively analyzed. In addition, the image recognition algorithm based on deep learning is further developed, which effectively avoids the limitation of missing proteins with irregular melting curves in the analysis process of conventional TPP methods [52]. Additionally, TPP was assisted with microspheres to enrich and precipitate proteins to simplify the sample pretreatment process and avoid sample loss as much as possible [53].
Currently, the mechanism of most antimalarial drugs in clinical remains elusive. CETSA-MS method was used to identify the targets of two important antimalarial drugs, quinine and mefloquine. Using Plasmodium falciparum lysate-infected erythrocytes, purine nucleoside phosphorylase (PfPNP) was identified as the common binding target of these two quinoline drugs, which was consistent with previous studies [54]. This is of great significance for new antimalarial drug research and developed, which aims at dealing with artemisinin resistance. Furthermore, TPP was applied to identify the target of the antihistamine cleomatine as Plasmodium chaperonin protein TRiC/CCT, thus explaining the mechanism of cleomatine in inhibiting multiple stages of parasite invasion [55]. Vioprolides A–D have significant effects against human acute lymphoblastic leukemia (ALL) cells, among which vioprolide A was deciphered as the most active vioprolide derivative, and nucleolar protein 14 (NOP14) was identified as a specific target of VioA [56]. Given that NOP14 was essential for ribosomal biogenesis and had been validated by cell activity assay, VioA showed unique anticancer therapeutic potential. Although these methods have been successfully used in numerous of drug identification cases, they are unlikely to be applied to those proteins highly inhomogeneous or displaying no curve shift upon drug binding. For example, some proteins will not aggregate after the unfolding of the ligand-binding domain [29] and some are stabilized by other proteins thus not affected by the drug binding [46]. Moreover, proteins those are resistant or not sensitive to temperature increment would not be captured by CETSA&TPP method.
Proteins denature in the presence of denaturants or organic solvents. In general, drug-bound proteins enhance antioxidant capacity while producing less precipitation in the presence of the same chemical agent compared with the drug-free proteins. Based on these properties, the following methods were established.
SPROX mainly focuses on the reduction of methionine oxidation rate upon drug binding, which proposes that the binding effect could increase the antioxidant capacity of the target [57, 58]. The protein mixtures with or without drug treatment are aliquoted in SPROX operation, and then different concentrations of denaturant (for example, guanidine hydrochloride or urea) and the same amount of hydrogen peroxide (H2O2) are added to oxidize the exposed methionine residues. To ensure a selective oxidization of methionine residues into sulfoxides, the concentration of H2O2 and reaction time should be optimized. The oxidation reaction is then quenched with excess methionine or catalase, and the reaction products are subjected to enzymatic digestion and LC-MS/MS analysis [28, 59]. As the amount of denaturing agent increases, proteins become more unfolded and more methionine-containing peptides will be oxidized. The relative quantification between methionine-containing peptides and their oxidized products is plotted as a function of the denaturant concentration. When half of the methionine-containing peptides are oxidized, the corresponding concentration of the denaturant is determined as the transition midpoint. The target protein could be pinpointed through the comparison of transition midpoint difference in the presence or absence of the drug. The developed SPROX is high throughput, which can simultaneously measure hundreds of proteins within a single test among complex biological samples [28].
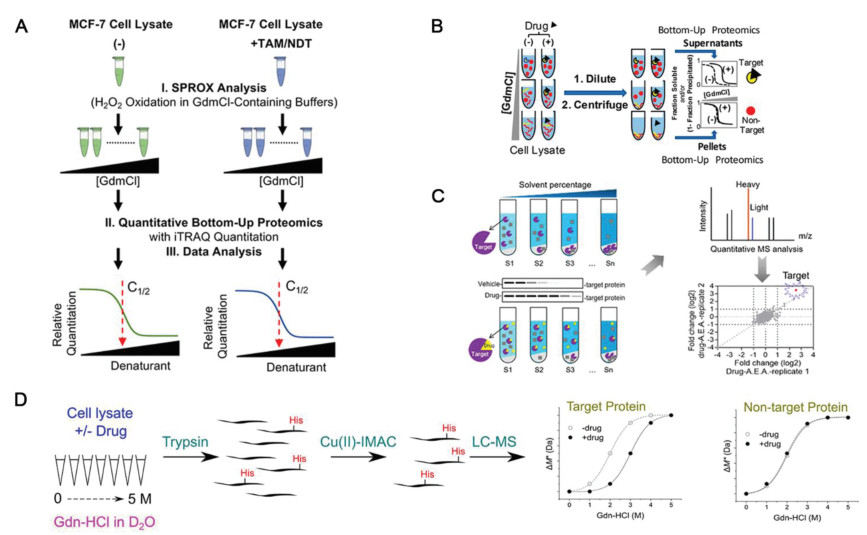
SPROX was further combined with the quantitative proteomic platform to explore the target proteins of the biologically active ligand resveratrol. In addition to the known cytosolic aldehyde dehydrogenase, six other potential new protein targets were revealed, including four targets related to the mechanism of protein translation [60]. Interestingly, eIF4A, which was previously detected by DARTS was not emerged as a potential target by SPROX, which might be attributed to its relative low binding affinity to resveratrol. In addition, the combined technologies of SILAC-SPROX (Stable isotope labeling by amino acids-SPROX) and iTRAQ-SPROX were used to explore targets of tamoxifen (TAM) and N-desmethyl tamoxifen (NDT) in the MCF-7 cell line. The stability change of more than 1000 proteins were analyzed, according to which 163 and 200 target proteins of TAM and NDT were identified, respectively. Among them, 11 and 20 of the identified targets had high reliability and had a high overlap rate, most of which were related to breast cancer (Fig. 4A) [61]. However, it is obvious that the application of SPROX is limited to the quantification of targets whose binding domains containing methionine. In addition, the efficiency of oxidation needs to be optimized, otherwise the distinction of the curve before and after drug binding will be affected.
In recent years, novel technologies have emerged for target discovery and identification. In 2018, Meng et al. established CPP, which facilitated both the drug target identification and the binding affinity determination (Fig. 4B) [62]. Similar to SPROX, CPP involved chemical denaturants, such as guanidine hydrochloride, to induce protein precipitation, which could be separated from the system via high-speed centrifugation. The retained soluble proteins were subsequently analyzed by LC-MS to obtain protein identities and the denaturation curves. Proteins those binding to the drug were supposed to have increased resistance to denaturants and thereby a shift for denaturation curve should be detected. Herein, CPP could significantly improve the protein coverage compared with that of SPROX while greatly reduce the false discovery rate. Furthermore, CPP also enlarges the detection to indirect protein targets of drugs, i.e drug-induced protein interactions, but only when the concentrations of proteins and ligands are sufficiently high.
In 2020, an energetics-based proteomics strategy, termed as SIP, was developed to analyze the interaction between drugs and their target proteins involving quantitative proteomics (Fig. 4C). Upon the addition of organic solvent at different proportions, such as acetone: ethanol: acetic acid = 50:50:0.1 (v: v: v), proteins underwent denaturation and formed precipitates due to the decrement of the solutions' dielectric constant and the destruction of the protein hydration membrane. The remained soluble proteins were analyzed by LC-MS/MS replication, and only proteins detected in two runs were subjected to further quantitative analysis, and the average value of log2 fold change (i.e. H/L normalized ratio) > 1 or < −1 was considered as a potential protein target [63]. Although the ratio and composition of organic reagents need to be further optimized, this method has the advantages of good practicability, universality and high throughput.
Both CPP and SIP have been validated with well-studied drugs and their targets, but limitations still remain: drug target identification by both methods rely on protein precipitation with denaturants, which may not cover all proteins, especially those with denaturant resistance, and the ratio of organic solvents should be carefully optimized for biological systems.
It can be seen it is a common problem for unmodified methods to explore target proteins with low abundance, which is proposed to be solved by His-HDX based method [64]. In this method, His-HDX was used as a readout technique for drug-induced protein stability variation monitoring (Fig. 4D). The workflow involves incubation of cell lysates in varying concentrations of protein denaturants in D2O with or without the addition of drug, followed up with enzymatic digestion, enrichment of histidine-containing peptides, and finally analyzed by LC-MS/MS. This method has been successfully applied to identify the interaction between endogenous MAPK14 and its inhibitor doramapimod in HEK293 cell lysates. This method has three significant initiatives: (1) Enrichment of histidine-containing peptides increases the likelihood of recognition of low abundance targets. A vital problem in the bottom-up proteomics approach is that the analytical capacity of LC-MS/MS measurements is limited, resulting in peptides from low-abundance proteins going undetected. The enrichment step will increase proteome coverage and thus increase the sensitivity of target identification; (2) Revealing drug binding site or solvent accessibility variation of target protein upon drug binding at amino acid-level; (3) Co-elution of deuterated and non-deuterated peptides in LC-MS/MS analysis; Therefore, it can intuitively calculate the fraction content of denatured and undenatured protein species in the sample using a single mass spectrometry rather than quantification in different samples or mass spectrometry. However, the experimental process is time-consuming, incubation with D2O alone takes ten days. Meanwhile, back exchange can lead to inaccurate calculation of hydrogen-deuterium exchange rate, thus affecting the accuracy of target identification. Moreover, for proteins with only a small amount of histidine, peptides without histidine will be lost after immobilized metal-ion affinity chromatography (IMAC) enrichment, resulting in difficult recognition of subsequent peptide mass fingerprint.
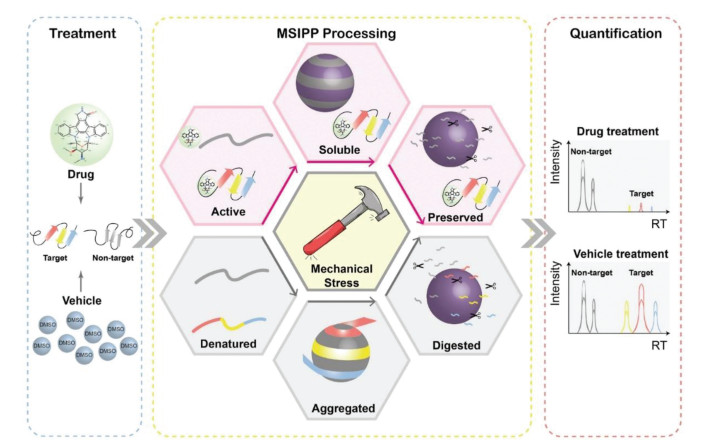
In 2021, a novel drug target screening method based on mechanical stress induced protein precipitation was developed (Fig. 5) [65]. As drug-bound proteins are more resistant to mechanical stress, they precipitate less than drug-free controls under the same mechanical stress. After the protein extract was treated with pharmaceutical compounds or solvents, the particles were mixed with the protein solution in a mass ratio at 2:1, and mechanical stress was generated by eddying the mixture. Protein precipitates caused by mechanical stress captured by particles were analyzed by mass spectrometry to screen potential targets. The MSIPP method successfully identified four clinical drugs with known targets including methotrexate (MTX), raltetrexate (RTX), SHP099 and geldamycin (GA), and demonstrated that DHFR is a binding target of RTX, which has not been revealed by any other unmodified method. Unlike protein denaturation due to heat or chemical forces, which is isotropic, mechanical forces may cause the protein to denature in certain physical directions, thus detecting interactions that cannot be revealed by these isotropic forces. However, the MSIPP method cannot be applied to living cells, and strong detergents such as SDS cannot be applied. Hence, some extremely hydrophobic membrane proteins will not be suitable. In addition, some proteins are easily captured by particles, while others are naturally stubborn and require more mechanical stress to precipitate. Moreover, different laboratories use different machines to provide mechanical stress, the parameters of precipitation caused by mechanical stress should be optimized, which is difficult to standardize.
Drug target discovery is a critical step for elucidating the mechanism of drug action. The popularization and development of methodology strongly support for target identification [66], upon which increasing number of drug targets were uncovered through various methods. We can also see from the examples that searching for potential drug targets by above methods is only one of many links, and the identification of targets requires the assistance of multiple means. Even each method still has own limitations, the combination of methods with different principles can obtain complimentary information (Table 1).
 DownLoad:
CSV
DownLoad:
CSV

|
In general, conventional modified-based methods usually miss target proteins with low affinities, and cannot effectively exclude the protein with non-specific binding, which usually lead to false positive results. What is more, drugs have to be chemically derivable and bioactivity verification is required before analysis. Unmodified methods can make up for the above limitations, according to which target proteins could be identified without chemical modification. Among them, DARTS and TPP are most widely used due to their relatively simple operation and high throughput. However, target proteins with low abundance would be missed due to the sensitivity limitation of the method, which asks for novel methods with improved detection sensitivity and specificity.
Besides, target screening by unmodified methods is greatly rely on the variations of physical and chemical properties of proteins, which cannot reach the targets with no such changes or lead to false negative results. It is important to note that target protein information obtained from each strategy is potential or putative, thus, further verified is still required. While look at it another way, there are still some properties of proteins that may wait to be developed into new methods. For example, proteins will denature under ultraviolet light, strong acid or strong alkali conditions, so whether proteins will have stronger tolerance to these external stimuli after combining with drugs remains to be further studied.
Actually, as an excellent target identification method, it should meet the following requirements: (1) the experimental conditions are easy to control and standardize. Although the properties of each drug and its targets are different, conditions such as incubation time, temperature and enzyme concentration may not be completely the same in the process of method optimization, but it is better to minimize such uncontrollable factors; (2) Retain the drug activity, and preferably apply the method to living cells or even in vivo, rather than confined to lysates, thus allowing for a more realistic simulation of the drug binding to the target; (3) To reduce costs. Drug research and development is a hard slog, and each link should be as economical as possible. Only in this way, more researchers can invest in the link of target search and lay a foundation for subsequent research. With the development of proteomics and mass spectrometry, increasing number of new technologies are emerged by integrating the advantages of different tactics while reduce the cost and time of detection. These new technologies will not only be applied to target identification of drugs, but also clarify the mechanism and toxicity of drugs much more clearly, providing an important method basis for solving difficult problems such as new drug development, drug mechanism research, and human disease marker identification.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by grants from the National Natural Science Foundation of China (No. 31870946), the Funding of Double First-rate discipline construction (No. CPU2018GF07), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Open Project Program of MOE Key Laboratory of Drug Quality Control and Pharmacovigilance (No. DQCP20/21MS01).
B. Raymer, S.K. Bhattacharya, J. Med. Chem. 61 (2018) 10375–10384. doi: 10.1021/acs.jmedchem.8b00407
S.J. Dixon, B.R. Stockwell, Curr. Opin. Chem. Biol. 13 (2009) 549–555. doi: 10.1016/j.cbpa.2009.08.003
J.G. Moffat, F. Vincent, J.A. Lee, J. Eder, M. Prunotto, Nat. Rev. Drug Discov. 16 (2017) 531–543. doi: 10.1038/nrd.2017.111
S. Sakamoto, Y. Kabe, M. Hatakeyama, Y. Yamaguchi, H. Handa, Chem. Rec. 9 (2009) 66–85. doi: 10.1002/tcr.20170
S. Sadegh, J. Matschinske, D.B. Blumenthal, Nat. Commun. 11 (2020) 3518. doi: 10.1038/s41467-020-17189-2
J. Vamathevan, D. Clark, P. Czodrowski, Nat. Rev. Drug Discov. 18 (2019) 463–477. doi: 10.1038/s41573-019-0024-5
C.L. Gao, G.G. Hou, J. Liu, Angew. Chem. Int. Ed. 59 (2020) 2429–2439. doi: 10.1002/anie.201912489
G.V. Paolini, R.H.B. Shapland, W.P. van Hoorn, J.S. Mason, A.L. Hopkins, Nat. Biotechnol. 24 (2006) 805–815. doi: 10.1038/nbt1228
X. Chen, Y. Wang, N. Ma, Signal Transduct. Targeted Ther. 5 (2020) 72. doi: 10.1117/12.2575904
A.C.M. van Esbroeck, A.P.A. Janssen, A.B. Cognetta, Science 356 (2017) 1084–1087. doi: 10.1126/science.aaf7497
Z. Slava, P. Verena, H. Christian, W. Herbert, Angew. Chem. Int. Ed. 52 (2013) 2744–2792. doi: 10.1002/anie.201208749
J. Du, J. Guo, D. Kang, Chin. Chem. Lett. 31 (2020) 1695–1708. doi: 10.1016/j.cclet.2020.03.028
Z. Yuan, M. Chen, J. Wang, Fish Shellfish Immunol. 79 (2018) 130–139. doi: 10.1016/j.fsi.2018.05.003
D.E. Gordon, G.M. Jang, M. Bouhaddou, Nature 583 (2020) 459–468. doi: 10.1038/s41586-020-2286-9
H. Deng, Q. Lei, Y. Wu, Y. He, W. Li, Eur. J. Med. Chem. 191 (2020) 112151. doi: 10.1016/j.ejmech.2020.112151
J. Xu, X. Li, K. Ding, Z. Li, Chem. Asian J. 15 (2020) 34–41. doi: 10.1002/asia.201901500
J. Wang, Q. Chen, Y. Shan, X. Pan, J. Zhang, Trends Anal. Chem. 115 (2019) 110–120. doi: 10.1016/j.trac.2019.03.028
L. Li, Y. Zhao, R. Cao, Chem. Commun. 55 (2019) 4407–4410. doi: 10.1039/c9cc00917e
J. Huang, H. Zhu, S.J. Haggarty, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 16594–16599. doi: 10.1073/pnas.0407117101
D. Xia, B. Liu, X. Xu, Y. Ding, Q. Zheng, J. Pharm. Anal. 11 (2021) 121–127.
X. Tu, X. Tan, X. Qi, J. Nanobiotechnol. 18 (2020) 32. doi: 10.1186/s12951-020-00591-9
B.L. Liu, H.C. Zhang, Y. Ding, Chin. Chem. Lett. 29 (2018) 1725–1730. doi: 10.1016/j.cclet.2018.12.006
Z.Q. Shi, Q.Q. Li, L. Mei, Chin. Chem. Lett. 31 (2020) 1345–1356. doi: 10.1016/j.cclet.2020.03.001
W.J. Wang, J. Wang, Y. Ding, J. Mater. Chem. B 8 (2020) 4813–4830. doi: 10.1039/c9tb02924a
J. Chang, Y. Kim, H.J. Kwon, Nat. Prod. Rep. 33 (2016) 719–730. doi: 10.1039/C5NP00107B
B. Lomenick, R. Hao, N. Jonai, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 21984–21989. doi: 10.1073/pnas.0910040106
M. Pepelnjak, N. de Souza, P. Picotti, Trends Biochem. Sci. 45 (2020) 919–920. doi: 10.1016/j.tibs.2020.05.006
E.C. Strickland, M.A. Geer, D.T. Tran, Nat. Protoc. 8 (2013) 148–161. doi: 10.1038/nprot.2012.146
D.M. Molina, R. Jafari, M. Ignatushchenko, Science 341 (2013) 84–87. doi: 10.1126/science.1233606
B. Lomenick, R.W. Olsen, J. Huang, ACS Chem. Biol. 6 (2011) 34–46. doi: 10.1021/cb100294v
J. Huang, B.E. Lomenick, R. Hao, Patent, US8703438 (B2), 2014.
F. Dal Piaz, M.B. Vera Saltos, S. Franceschelli, et al., J. Nat. Prod. 79 (2016) 2681–2692. doi: 10.1021/acs.jnatprod.6b00627
Q. Li, L. Cao, Y. Tian, Mol. Cell. Proteom. 17 (2018) 1531–1545. doi: 10.1074/mcp.ra118.000752
B. Lomenick, G. Jung, J.A. Wohlschlegel, J. Huang, Curr. Protoc. Cell Biol. 3 (2011) 163–180. doi: 10.1002/9780470559277.ch110180
R. Su, L. Dong, C. Li, Cell 172 (2018) 90–105. doi: 10.1016/j.cell.2017.11.031
J.J. Chen, R. Su, Patent, US11179360 (B2), 2021.
Y. Huang, R. Su, Y. Sheng, Cancer Cell 35 (2019) 677–691. doi: 10.1016/j.ccell.2019.03.006
R. Su, L. Dong, Y. Li, Cancer Cell 38 (2020) 79–96. doi: 10.1016/j.ccell.2020.04.017
Y. Yang, L. Cao, H. Gao, J. Med. Chem. 62 (2019) 4056–4073. doi: 10.1021/acs.jmedchem.9b00091
D. Minond, G.B. Fields, M. Giulianotti, Patent, US11149028 (B2), 2021.
P. Leuenberger, S. Ganscha, A. Kahraman, Science 355 (2017) eaai7825. doi: 10.1126/science.aai7825
Y. Feng, G. De Franceschi, A. Kahraman, Nat. Biotechnol. 32 (2014) 1036–1044. doi: 10.1038/nbt.2999
I. Piazza, K. Kochanowski, V. Cappelletti, Cell 172 (2018) 358–372. doi: 10.1016/j.cell.2017.12.006
S. Schopper, A. Kahraman, P. Leuenberger, Nat. Protoc. 12 (2017) 2391–2410. doi: 10.1038/nprot.2017.100
P. Nordlund, Patent, US9528996 (B2), 2016.
T. Friman, Biorg. Med. Chem. 28 (2020) 115174. doi: 10.1016/j.bmc.2019.115174
T.P. Miettinen, M. Bjorklund, Mol. Pharm. 11 (2014) 4395–4404. doi: 10.1021/mp5004866
T. Ishii, T. Okai, M. Iwatani-Yoshihara, Sci. Rep. 7 (2017) 13000. doi: 10.1038/s41598-017-12513-1
M.M. Savitski, F.B.M. Reinhard, H. Franken, Science 346 (2014) 1255784. doi: 10.1126/science.1255784
H. Franken, T. Mathieson, D. Childs, Nat. Protoc. 10 (2015) 1567–1593. doi: 10.1038/nprot.2015.101
F.B.M. Reinhard, D. Eberhard, T. Werner, Nat. Methods 12 (2015) 1129–1134. doi: 10.1038/nmeth.3652
C. Ruan, W. Ning, Z. Liu, ACS Chem. Biol. 17 (2022) 252–262. doi: 10.1021/acschembio.1c00936
M.L. Ye, J.W. Lv, Patent, CN114076826 (A), 2022.
J. Dziekan, H. Yu, D. Chen, Sci. Transl. Med. 11 (2019) eaau3174. doi: 10.1126/scitranslmed.aau3174
K.Y. Lu, B. Quan, K. Sylvester, Proc. Natl. Acad. Sci. U. S. A. 117 (2020) 5810–5817. doi: 10.1073/pnas.1913525117
V.C. Kirsch, C. Orgler, S. Braig, Angew. Chem. Int. Ed. 59 (2020) 1595–1600. doi: 10.1002/anie.201911158
G.M. West, L. Tang, M.C. Fitzgerald, Anal. Chem. 80 (2008) 4175–4185. doi: 10.1021/ac702610a
E.J. Walker, J.Q. Bettinger, K.A. Welle, J.R. Hryhorenko, S. Ghaemmaghami, Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 6081–6090. doi: 10.1073/pnas.1819851116
A. Cabrera, N. Wiebelhaus, B. Quan, J. Am. Soc. Mass Spectrom. 31 (2020) 217–226. doi: 10.1021/jasms.9b00041
P.D. Dearmond, Y. Xu, E.C. Strickland, K.G. Daniels, M.C. Fitzgerald, J. Proteome Res. 10 (2011) 4948–4958. doi: 10.1021/pr200403c
R.N. Ogburn, L. Jin, H. Meng, M.C. Fitzgerald, J. Proteome Res. 16 (2017) 4073–4085. doi: 10.1021/acs.jproteome.7b00442
H. Meng, R. Ma, M.C. Fitzgerald, Anal. Chem. 90 (2018) 9249–9255. doi: 10.1021/acs.analchem.8b01772
X. Zhang, Q. Wang, Y. Li, Anal. Chem. 92 (2020) 1363–1371. doi: 10.1021/acs.analchem.9b04531
M. Miyagi, K. Tanaka, S. Watanabe, J. Kondo, T. Kishimoto, Anal. Chem. 93 (2021) 14985–14995. doi: 10.1021/acs.analchem.1c02283
J. Lyu, Y. Wang, C. Ruan, Anal. Chim. Acta 1168 (2021) 338612. doi: 10.1016/j.aca.2021.338612
H.Y. Hwang, T.Y. Kim, M.A. Szasz, Proteomics 20 (2020) e1900325. doi: 10.1002/pmic.201900325

Figure 1 An illustration of main unmodified methodologies for identifying and validating drug targets. The gray sections represent new methods to be developed.

Figure 2 (A) Schematic illustration of working mechanism of DARTS. Reproduced with permission [30]. Copyright 2010, American Chemical Society. (B) The working process of DARTS. (C) Incubated with indicated drug (rapamycin or FK506), FKBP12 was difficult to be digested by subtilisin. (D) The chemical structure of resveratrol and thermolysin digestion result of yeast cell lysates and human HeLa cell lysates treated with resveratrol. Reproduced with permission [26]. Copyright 2009, National Academy of Sciences. (E) Experimental LiP-MS workflow. Red pentagon, drug; K, lysine residue; R, arginine residue; Rel. int., relative intensity.

Figure 3 (A) Western blot analysis for isolated spleen. The drug-bound protein had more obvious bands in the supernatant, and the total protein content was almost unchanged. Reproduced with permission [48]. Copyright 2017, Ishii et al. (B) Quantitative proteome-wide profiling of protein thermal stability. (1) Cells were cultured under differential conditions, with (+) or without (−) the drug. (2) The sample was divided into aliquots (usually are 10 samples, as TMT can label up to 10 samples). (3) Aliquots were subjected to heating at the indicated temperatures. (4) After digestion with trypsin, each sample was labeled. (5) Subsequently, all samples from each condition were mixed and analyzed using LC-MS/MS. The obtained reporter ion intensities were used to fit a melting curve and calculate the melting temperature Tm of each protein separately for the two conditions. The target protein melt curve can show a significant shift upon drug binding.

Figure 4 (A) Schematic representation of the iTRAQ-SPROX experimental workflow applied in this work. Reproduced with permission [61]. Copyright 2017, American Chemical Society. (B) Schematic representation of the chemical denaturation and protein precipitation (CPP). Reproduced with permission [62]. Copyright 2018, American Chemical Society. (C) Organic solvent-induced protein precipitation (SIP). Reproduced with permission [63]. Copyright 2020, American Chemical Society. (D) Histidine hydrogen-deuterium exchange (His-HDX). Reproduced with permission [64]. Copyright 2021, American Chemical Society.

Figure 5 Workflow of MSIPP. After drug combination or vehicle treatment, the entire proteome of both treatments is forced to precipitate under mechanical stress. Drug-bound proteins are more resistant to precipitation than free proteins, resulting in less concentration of drug-binding proteins on the particles. Finally, quantitative proteomics can reveal differences in target proteins in drug/vehicle therapy. Reproduced with permission [65]. Copyright 2021, Elsevier B.V.
Table 1. The advantages and limitations of various unmodified target discovery methodologies.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们