Citation:
Sumaira Nazar Hussain, Yana Men, Zhen Li, Pingping Zhao, Gongzhen Cheng, Wei Luo. Molybdenum-induced tuning 3d-orbital electron filling degree of CoSe2 for alkaline hydrogen and oxygen evolution reactions[J]. Chinese Chemical Letters,
2023, 34(2): 107364.
doi:
10.1016/j.cclet.2022.03.087
Molybdenum-induced tuning 3d-orbital electron filling degree of CoSe2 for alkaline hydrogen and oxygen evolution reactions
English
Molybdenum-induced tuning 3d-orbital electron filling degree of CoSe2 for alkaline hydrogen and oxygen evolution reactions
wluo@whu.edu.cn (W. Luo). 1 These authors contributed equally to this work.
Received Date:
16 December 2021 Accepted Date:
22 March 2022 Revised Date:
09 February 2022 Available Online:
15 February 2023
Abstract:
The development of high-performance non-precious metal-based robust bifunctional electrocatalyst for both hydrogen evolution reaction (HER) and oxygen evolution reactions (OER) in alkaline media is essential for the electrochemical overall water splitting technologies. Herein, we demonstrate that the HER/OER performance of CoSe2 can be significantly enhanced by tuning the 3d-orbital electron filling degree through Mo doping. Both density functional theory (DFT) calculations and experimental results imply that the doping of Mo with higher proportion of the unoccupied d-orbital (Pun) could not only serve as the active center for water adsorption to enhance the water molecule activation, but also modulate the electronic structures of Co metal center leading to the optimized adsorption strength of *H. As expected, the obtained Mo-CoSe2 exhibits a remarkable bifunctional performance with overpotential of only 85 mV for HER and 245 mV for OER to achieve the current density of 10 mA/cm2 in alkaline media. This work will provide a valuable insight to design highly efficient bifunctional electrocatalyst towards HER and OER.
Electrochemical water splitting powered by renewable and clean energy sources has been widely proposed to be the most promising, cleanest and efficient technology for large scale hydrogen production, which plays a crucial role in "hydrogen economy" [1-9]. Specially, the alkaline water electrolysis has attracted increasing attention owing to the development of low-cost catalysts utilized for anodic oxygen evolution reaction (OER) with benchmark Ir/Ru performance in alkaline electrolytes [10-15]. Nevertheless, for cathode hydrogen evolution reaction (HER), the kinetics of HER often decreases by about 2 orders of magnitude than that in acidic condition even when it is catalyzed by the state-of-the-art Pt catalyst [16-19]. More critically, the high cost and natural scarcity of the most efficient Pt-based HER catalyst or Ir-based OER catalyst have seriously hindered the practical application of water electrolysis [20-24]. Therefore, it is highly desirable and challenging to design highly efficient and low-cost bifunctional catalysts driving OER and HER simultaneously in alkaline media, which is of great significance for the large-scale application and development of viable alkaline water electrolytic system.
Recently, great efforts have been devoted to developing the cost-effective transition metal-based bifunctional catalysts for water splitting as alternatives to the noble metal catalysts [25] such as nitrides [26] carbides [27] oxides [28] selenides [29] phosphides [30, 31] and sulphides [32]. Among them, transition metal selenides have attracted a great deal of attentions due to their intrinsic advantages of superior conductivity and chemical composition tenability [33]. Particularly, cobalt selenide (CoSe2), where the Co 3d electron adopts a low spin electron configuration of t2g6 eg1, is considered as a promising bifunctional catalyst, due to its properties of a metallic conductor [34, 35]. Although highly efficient CoSe2 based catalysts driving OER have been widely reported in alkaline media, their alkaline HER catalytic activities are still far lower than the state-of-the-art Pt/C, which is possibly derived from the sluggish water adsorption/dissociation and the strong hydrogen binding strength.
Herein, we report a facile synthesis strategy to simultaneously boost the bifunctional catalytic activity of CoSe2 by introducing molybdenum (Mo) with higher proportion of unoccupied d-orbitals (Pun). Density functional theory (DFT) calculations further reveal that the doped Mo with higher Pun endows the CoSe2 with dual activity sites, in which the Mo with higher Pun could act as the oxophilic sites to strengthen the interaction between water molecule and metal center, and tailor the electronic structure of Co atom, leading to the optimized H adsorption, and enhanced alkaline HER performance. Furthermore, benefiting from the introduction of oxyphilic molybdenum atoms, the interaction between the catalytic center and various oxygen-containing intermediates could be optimized, which will productively improve the OER performance. As expected, the obtained Mo doped CoSe2 (Mo-CoSe2) possesses an excellent catalytic activity for both HER and OER in alkaline media, with only 85 mV and 245 mV to achieve the current density of 10 mA/cm2, respectively, which are much better than most of recently reported transition metal selenide-based catalysts for HER and even better than IrO2 catalyst for OER.
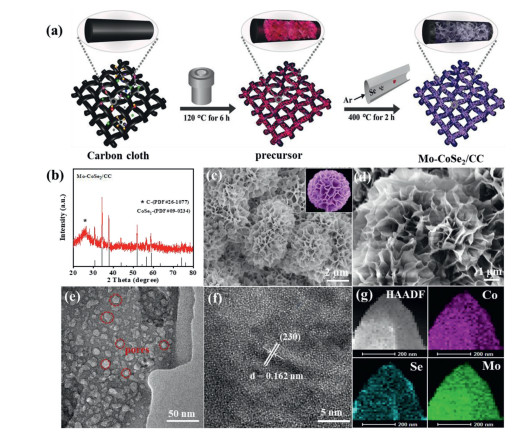
As schematically illustrated in Fig. 1a, Mo doped CoSe2 nanosheets supported on carbon cloth (Mo-CoSe2/CC) were successfully fabricated by growing Mo doped Co(OH)F/CC on flexible carbon cloth (CC) via a facile hydrothermal method followed by a subsequent annealing treatment with selenium powder under argon gas flow. The crystalline structure of precursor was investigated by X-ray diffraction spectroscopy (XRD). As shown in Fig. S1a (Supporting information), a series of characteristic diffraction peaks match well with the standard Co(OH)F crystalline phase (JCPDS #50-0827). Typical scanning electron microscopy (SEM) image of Mo-Co(OH)F/CC (Fig. S1b in Supporting information) display layered nanosheets morphology with a flower-like architecture. After selenylation, the XRD pattern (Fig. 1b) of Mo-CoSe2/CC shows the characteristic diffraction peaks of CoSe2 crystalline phase (JCPDS #09-0234), indicating the successful conversion from Co(OH)F to CoSe2. Meanwhile, the morphology structure of the flower-like nanosheets of the Mo-CoSe2/CC are maintained well (Figs. 1c and d). Transmission electron microscopy (TEM) images further reveal the nanosheet morphology of the Mo-CoSe2/CC. It can be seen clearly that a large number of porous structures with abundant edges appear on the nanosheet (Fig. 1e), which contributes to expose more active sites and accelerate the mass transfer [36, 37]. Furthermore, the high-resolution transmission image (HRTEM) shows the identified lattice spacing of 0.162 nm, which is assigned to the (230) planes of CoSe2 (Fig. 1f). The high annular dark-field scanning TEM (HAADF-STEM) and corresponding elemental mapping images (Fig. 1g) of the Mo-CoSe2/CC incontestably demonstrate the homogeneously spatial distribution of Mo, Co, and Se throughout the catalyst. For comparison, pure CoSe2 catalyst was also prepared by similar synthetic method. The XRD pattern (Fig. S2a in Supporting information) and SEM image (Figs. S2b in Supporting information) indicate that the CoSe2 catalyst possesses the same crystal phase and nanoarray morphology structure. In addition, the full-width at half-maxima (FWHM) analysis (Fig. S3 in Supporting information) shows the higher disorder in the Mo-CoSe2 than that of pure CoSe2 crystalline phase, which is probably derived from lattice distortions of CoSe2 caused by the incorporation of Mo-Se bond [38-40]. In addition, the Mo-CoSe2/CC nanosheets with different Mo doping contents were also prepared, and characterized by XRD and SEM (Figs. S4 and S5 in Supporting information).
Figure 1
Figure 1.
(a) Schematics of the synthesis of Mo-CoSe2/CC nanosheets. (b) XRD pattern of Mo-CoSe2/CC. (c, d) Scanning electron microscopy (SEM) images of Mo-CoSe2/CC nanosheets. (e) Transmission electron microscopy (TEM) images of Mo-CoSe2/CC. (f) High-resolution transmission electron microscope (HRTEM) of Mo-CoSe2/CC. (g) High-angle annular dark-field (HAADF)-STEM image with corresponding element maps of Mo-CoSe2/CC.
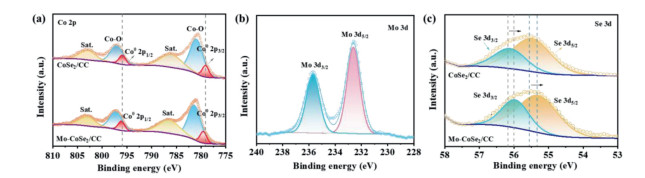
In order to investigate the chemical composition and valence state of catalysts, Mo-CoSe2/CC and CoSe2 were further analysed by X-ray photoelectron spectroscopy (XPS). As illustrated in Fig. 2a, a pair of typical characteristic peaks at the binding energies of 779.9 eV and 793.8 eV can be seen unambiguously in the Co 2p XPS spectrum of CoSe2, corresponding to Co0 2p3/2 and Co0 2p1/2, respectively [41]. The pair of shoulder peaks at the binding energies of 781.3 and 797.3 eV belonging to cobalt oxide might arise from surface oxidation exposed to air [42]. The peaks located at binding energies of 786.4 and 803.2 eV are assigned to relevant satellite peak [43]. Notably, after the introduction of molybdenum, slight positive shifts of 0.6 eV and 0.4 eV for Co0 2p3/2 and Co0 2p1/2 can be observed in the Co 2p XPS spectrum of Mo-CoSe2/CC compared with the pure CoSe2/CC. The XPS spectrum of Mo 3d core level (Fig. 2b) shows two main peaks of Mo 3d3/2 and Mo 3d5/2, indicating that Mo mainly exists in the form of Mo6+ [44, 45]. The Se 3d core level spectrum of Mo-CoSe2/CC (Fig. 2c) can be deconvoluted into two peaks with binding energies of 55.9 and 55.3 eV, assigned to Se 3d3/2 and Se 3d5/2 [46-48]. It can be seen clearly that the peaks of Se 3d3/2 and Se 3d5/2 of Mo-CoSe2/CC display negative shifts of 0.3 eV and 0.2 eV compared to those of CoSe2/CC, respectively. These shifts of binding energy indicate that the Mo doping induces electron transfer between cobalt and Se, which is possibly conducive to regulate the adsorption energy of reaction intermediates during the catalytic process, leading to the promoted reaction rate.
Figure 2
Figure 2.
(a) XPS spectra of Co 2p in the Mo-CoSe2/CC and CoSe2/CC. (b) XPS spectrum of Mo 3d of Mo-CoSe2/CC. (c) XPS spectra of Se 3d in the Mo-CoSe2/CC and CoSe2/CC.
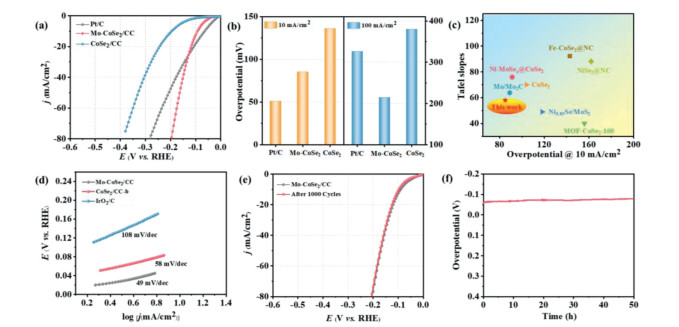
The HER performance of all the samples were investigated using a standard three electrode system in 1.0 mol/L KOH. As shown in Fig. S6 (Supporting information), among all the samples tested, the CoSe2/CC with 0.3 mmol of Mo doping (named Mo-CoSe2/CC) possesses the highest catalytic activity. Furthermore, the linear sweep voltammetry (LSV) curves (Figs. 3a and b) show that the Mo-CoSe2/CC displays superior HER activity with overpotential of only 85 mV and 214 mV to achieve the current density of 10 mA/cm2 and 100 mA/cm2, respectively, which are much better than that of CoSe2/CC (136 mV and 380 mV) and even comparable to Pt/C (51 mV and 326 mV). Moreover, the catalytic activity of Mo-CoSe2/CC is also higher than most of the recently reported transition metal selenides-based and other non-noble metal-based electrocatalysts toward alkaline HER (Fig. 3c and Table S1 in Supporting information). The Tafel slopes of samples have been assessed to evaluate the HER kinetics. Fig. 3d demonstrates the smaller Tafel slopes of Mo-CoSe2/CC (58 mV/dec) comparing with that of CoSe2/CC (108 mV/dec), indicating that the HER kinetics of CoSe2/CC is enhanced by Mo doping. Furthermore, the Tafel slope of Mo-CoSe2/CC indicates that the alkaline HER process catalyzed by Mo-CoSe2/CC undergoes the Volmer-Heyrovsky mechanism [49]. The electrochemical surface area (ECSA) of Mo-CoSe2/CC and CoSe2/CC were determined from corresponding electrochemical double-layer capacitances (Cdl) by a well-established cyclic voltammetry method (Fig. S7 in Supporting information).
Figure 3
Figure 3.
Electrocatalytic HER activities in 1 mol/L KOH. (a) LSV curves of Pt/C, CoSe2 and Mo-CoSe2 in 1.0 mol/L KOH. (b) The overpotential of Pt/C, CoSe2 and Mo-CoSe2 at 10 mA/cm2 and 100 mA/cm2, respectively. (c) Comparison of the alkaline HER performance of Mo-CoSe2 with other reported transition metal based electrocatalysts. (d) The Tafel slopes of Pt/C, CoSe2 and Mo-CoSe2 (e) LSV curves of Mo-CoSe2 before and after 1000 cycles. (f) The chronopotentiometric curve at the constant current density of 10 mA/cm2 for 50 h.
The corresponding higher Cdl value of Mo-CoSe2/CC (144.6 mF/cm2) comparing with that of CoSe2/CC (80.1 mF/cm2) reveals that more reactive active sites are exposed in Mo-CoSe2/CC, which contributes to the promoted HER kinetics. Meanwhile, electrochemical impedance spectroscopy (EIS) measurements were also performed to investigate the influence of Mo dopant on the interface charge transfer kinetics [50]. The Nyquist plots (Fig. S8 in Supporting information) illustrate that the Mo-CoSe2/CC exhibits the smaller polarization resistance at high frequency ranges than pure CoSe2, indicating that the charge transfer kinetics of CoSe2/CC can be accelerated effectually by Mo doping, which is beneficial to the enhanced HER performance. Furthermore, the stability of Mo-CoSe2/CC was evaluated. As shown in Fig. 3e, the LSV curve recorded after 1000 cycles is coincident with the initial polarization curve, highlighting the superior stability. Similarly, almost unchanged overpotential over 50 h (Fig. 3f) can be observed in the chronopotentiometric curve at the current density of 10 mA/cm2, further suggesting the good stability of Mo-CoSe2/CC. Moreover, SEM (Fig. S9 in Supporting information) and TEM images (Fig. S10 in Supporting information) after durability test show that the nanosheet architecture of Mo-CoSe2/CC is maintained well without structural destruction. Moreover, the XRD pattern (Fig. S11 in Supporting information) and XPS spectra (Fig. S12 in Supporting information) of Mo-CoSe2/CC after stability test reveal the stable CoSe2 crystalline phase and almost unchanged surface compositions, further indicating the good stability of Mo-CoSe2/CC toward alkaline HER.
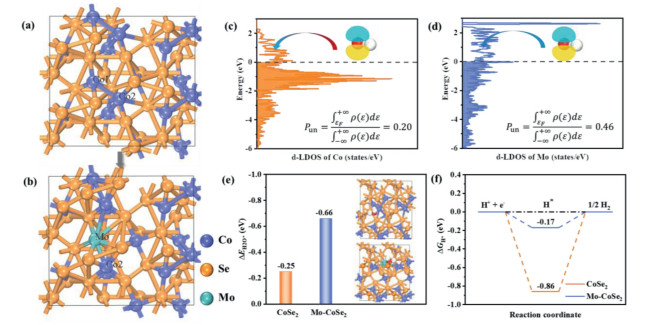
To reveal the enhancement mechanism of Mo-CoSe2 catalyst toward alkaline HER, a systematic investigation of HER process on CoSe2 and Mo-CoSe2 have been carried out by density functional theory (DFT) calculations. In order to reveal the influence of Mo dopant on the catalytic activity, we used one Mo atom to substitute one Co atom on the pristine CoSe2 surface. Two possible sites replaced by Mo atom are considered and Co1 site is finally selected as the stable substitution site, according to the results of calculated formation energies (Table S2 in Supporting information). The optimized structures of CoSe2 and Mo-CoSe2 are shown in Figs. 4a and b, respectively. The Pun of various metal sites in Mo-CoSe2 and CoSe2 are first investigated. As shown in Figs. 4c and d, the Pun of Mo atom in Mo-CoSe2 is calculated to be 0.46, which is higher than that of Co atom in pristine CoSe2 (~0.20). This result indicates that Mo atom could provide more unoccupied 3d-orbitals to accommodate the lone pair electrons of water molecule and thereby enhance the interaction in between, which contributes to the water activation and dissociation in the Volmer step.
Figure 4
Figure 4.
(a, b) Optimized CoSe2 (210) surface before and after Mo doping. Purple, orange and green spheres represent Co, Se and Mo atoms, respectively. (c) The d-orbital local DOS (d-LDOS) of Co atom in the pristine CoSe2. (d) The d-orbital local DOS (d-LDOS) of Mo atom in Mo-CoSe2. (e) Calculated free energy of H2O adsorption on CoSe2 and Mo-CoSe2 surface. The insets are the optimized structures with H2O adsorption. (f) Calculated free energy diagram of H adsorption on CoSe2 and Mo-CoSe2 surface.
Subsequently, we compared the binding strength of H2O on both catalysts and the optimized structures are shown in Fig. S13 (Supporting information). As expected, the Mo-CoSe2 possesses a more negative adsorption free energy of water (ΔG*H) than CoSe2 (Fig. 4e), implying that the water adsorption on the CoSe2 can be enhanced by Mo doping, leading to the acceleration of water dissociation process [51-54]. Furthermore, the adsorption free energy of hydrogen atom (ΔG*H) is also investigated, which is also a critical factor in evaluating the HER activities of electrocatalysts [55]. As shown in Fig. 4f, the ΔG*H after Mo substitution (Fig. S14 in Supporting information) is much more thermal neutral than that of pristine CoSe2, which is conducive to the promoted HER kinetics.
The OER performance of these samples was further evaluated in 1.0 mol/L KOH electrolyte. As expected, the Mo-CoSe2/CC with 0.3 mmol of Mo doping also exhibits the highest OER activity among the various CoSe2/CC with different Mo doping content (Fig. S15 in Supporting information). Subsequently, the OER activity of Mo-CoSe2/CC was also compared with that of benchmark IrO2 catalyst and pure CoSe2/CC. As shown in Figs. S16a and b (Supporting information), the Mo-CoSe2/CC requires the overpotentials of 245 mV and 390 mV to achieve the current density of 10 mA/cm2 and 100 mA/cm2, respectively, which are much lower than those of IrO2 catalyst (280 mV and 410 mV) and CoSe2/CC (320 mV and 480 mV). The superior catalytic activity of Mo-CoSe2/CC is also better than most of the recently reported transition metal selenides-based and other non-noble metal based electrocatalysts toward alkaline OER (Fig. S16c in Supporting information, Table S3). As shown in Fig. S16d (Supporting information), the smallest Tafel slope of Mo-CoSe2/CC among all the samples indicates the fastest OER reaction kinetics [56, 57]. Meanwhile, the Nyquist plots (Fig. S17 in Supporting information) indicate that the Mo-CoSe2/CC possesses the faster charge transfer kinetics than that of CoSe2/CC, leading to accelerated OER process [58, 59]. The ECSA was also evaluated by Cdl measurement [60]. As shown in Fig. S18 (Supporting information), the ECSA of Mo-CoSe2/CC is measured to be 116 mF/cm2 which is higher than that of the CoSe2 (73.4 mF/cm2), implying more active site exposure on the Mo-CoSe2/CC. The stability of Mo-CoSe2/CC was also investigated. As shown in Fig. S16e (Supporting information), after 1000 cycles of CV measurement, there is negligible degradation comparing with the first cycle. Meanwhile, almost no obvious current degradation is observed after 35 h of chronoamperometric test for the Mo-CoSe2/CC (Fig. S16f in Supporting information).
In summary, the Mo-CoSe2 bifunctional catalyst has been rational designed via a simple two-step synthetic method. DFT calculations demonstrate that the introdction of Mo dopant with a higher proportion of the unoccupied d orbital (Pun) could lead to enhanced water adsorption, and promoted Volmer step, which together with the optimized binding energy of H*, contributes to the enhanced alkaline HER performance, with 85 mV to achieve the current density of 10 mA/cm2. Moreover, the obtained Mo-CoSe2/CC catalysts also exhibit superior OER catalytic activity with 245 mV to achieve the current density of 10 mA/cm2 in 1 mol/L KOH. This design strategy though doping engineering by introducing of transition metal with higher Pun to promote the alkaline bifunctional catalytic activity for both HER and OER paves a new way to the rational design of other highly efficient multi-functional electrocatalysts.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No. 21972107), Natural Science Foundation of Jiangsu Province (No. BK20191186), and Natural Science Foundation of Hubei Province (No. 2020CFA095). The numerical calculations in this paper have been done on the supercomputing system in the Supercomputing Center of Wuhan University.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.087.
T. Meng, J.W. Qin, D. Xu, et al., ACS Appl. Mater. Interfaces 11 (2019) 9023–9032. doi: 10.1021/acsami.8b19341
[59]
H.J. Yan, Y. Xie, A.P. Wu, et al., Adv. Mater. 31 (2019) 1901174. doi: 10.1002/adma.201901174
[60]
S. Zhao, R.X. Jin, H. Abroshan, et al., J. Am. Chem. Soc. 139 (2017) 1077–1080. doi: 10.1021/jacs.6b12529
Figure 1
(a) Schematics of the synthesis of Mo-CoSe2/CC nanosheets. (b) XRD pattern of Mo-CoSe2/CC. (c, d) Scanning electron microscopy (SEM) images of Mo-CoSe2/CC nanosheets. (e) Transmission electron microscopy (TEM) images of Mo-CoSe2/CC. (f) High-resolution transmission electron microscope (HRTEM) of Mo-CoSe2/CC. (g) High-angle annular dark-field (HAADF)-STEM image with corresponding element maps of Mo-CoSe2/CC.
Figure 2
(a) XPS spectra of Co 2p in the Mo-CoSe2/CC and CoSe2/CC. (b) XPS spectrum of Mo 3d of Mo-CoSe2/CC. (c) XPS spectra of Se 3d in the Mo-CoSe2/CC and CoSe2/CC.
Figure 3
Electrocatalytic HER activities in 1 mol/L KOH. (a) LSV curves of Pt/C, CoSe2 and Mo-CoSe2 in 1.0 mol/L KOH. (b) The overpotential of Pt/C, CoSe2 and Mo-CoSe2 at 10 mA/cm2 and 100 mA/cm2, respectively. (c) Comparison of the alkaline HER performance of Mo-CoSe2 with other reported transition metal based electrocatalysts. (d) The Tafel slopes of Pt/C, CoSe2 and Mo-CoSe2 (e) LSV curves of Mo-CoSe2 before and after 1000 cycles. (f) The chronopotentiometric curve at the constant current density of 10 mA/cm2 for 50 h.
Figure 4
(a, b) Optimized CoSe2 (210) surface before and after Mo doping. Purple, orange and green spheres represent Co, Se and Mo atoms, respectively. (c) The d-orbital local DOS (d-LDOS) of Co atom in the pristine CoSe2. (d) The d-orbital local DOS (d-LDOS) of Mo atom in Mo-CoSe2. (e) Calculated free energy of H2O adsorption on CoSe2 and Mo-CoSe2 surface. The insets are the optimized structures with H2O adsorption. (f) Calculated free energy diagram of H adsorption on CoSe2 and Mo-CoSe2 surface.

 DownLoad:
DownLoad:

 下载:
下载:





