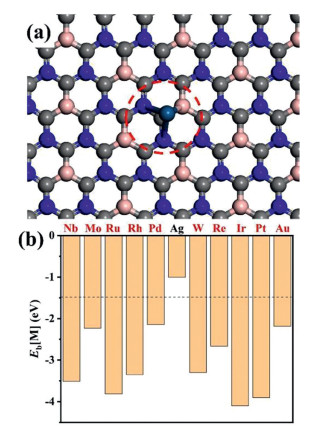
Figure 1.
(a) The configuration of 2D BC3N2 with metal atom loaded on it. The atom in the red cycle denotes the doped metal atom. (b) Binding energies of metal atoms on 2D BC3N2.
Computational design of BC3N2 based single atom catalyst for dramatic activation of inert CO2 and CH4 gasses into CH3COOH with ultralow CH4 dissociation barrier
Chenxu Zhao , Menghui Xi , Jinrong Huo , Chaozheng He , Ling Fu

The increased CO2 level in the atmosphere has caused many damage in climate, such as greenhouse effect. Therefore, the topic on carbon capture and storage has been proposed as a solution. The captured CO2 has also stimulated the research of CO2 conversion [1-3]. In the meantime, CH4 has been treated as an attractive source of hydrogen energy at present and can be obtained from coal-bed gas in large quantity [4]. The CO2 conversion with the assistance of CH4 can realize the synthesis of value-added chemicals such as HCOOH, CH3COOH, CH3OH, CO [5]. Among these products, the synthesis of CH3COOH has been investigated extensively due to the 100% utilization of atoms in CO2 and CH4. Besides, CH3COOH is a fuel with high energy density compared to other C1 products [6-9]. This strategy can alleviate human's dependence on conventional fossil fules and simultaneously reduce the detrimental impact of greenhouse gasses. The most widely studied reaction for simultaneous activation of CO2 and CH4 is the production of CO/H2 syngas [10-15]. However, the study of the coupling of CH4 and CO2 to C2 products is still an emerging area. The major challenge of this reaction exists in the simultaneous activation of CH4 and CO2, especially the process of CH4 dissociation into CH3 and H. These have been extensively studied in our previous works [16-19]. Therefore, it is highly desired to develop novel catalyst with high efficiency. As reported in previous studies, the final products of CO2~CH4 co-activation mainly include CH3COOH and HCOOH. The major intermediate of CH3COOH production is CH3COO, which derived from the combination between CH3 and CO2. For HCOOH production, there exists two intermediates: COOH and HCOO. In other words, the H atom can bond with CO2 on both C and O atoms. In pioneering works, direct production of CH3COOH via CO2 and CH4 has been carried out on several heterogeneous catalysts, such as Co-Cu metal oxides [7], zeolite based catalysts [20], (ZnO)3-In2O3 [21], Pd/C [8], Pt/Al2O3 [9], Co-Pd/TiO2 [22], NiO/NiF [23] and ZnO/r-GO [24]. While, the low conversion efficiency and poor selectivity still hinder the development of catalysts in this direction. The main issue that hinders the reactivity of catalysts exists in the effective adsorption and dissociation of inert CH4. Wang et al. have obtained only −0.04 eV adsorption energy for CH4 on Co(001) surface, which agrees with the following study of Zhang et al. (−0.06 eV) [25, 26]. Besides, The activation barrier for CH4 dehydrogenation, *CH4 → *CH3 + *H, has reached to 0.95 eV in Zhang's study [26]. Deo et al. have also revealed the weak van der Waals interaction between CH4 and Ni based alloys with only ~−0.02 eV adsorption energies [27]. Moreover, the reaction barrier of CH4 dissociation has reached to 1.78 eV (0.92 eV) for Cu (Ni) [27].
In order to improve the reactivity, many research has also focused on the reaction mechanism. The first step of coupling between CO2 and CH4 is CH4 adsorption and further dehydrogenation to CH3, which has been extensively studied on metals [21, 28-36]. Ge et al. have realized the C-H activation in CH4 via introducing M-O pair at oxide-oxide interface [21]. Hu et al. has studied the transformation of CH4 to C and H on Pd(100) surface from the aspect of valencies, which emerges clear relationship with the location of transition states [30]. Kim et al. have introduced OH groups onto Ir(110) surface and successfully achieved CH4 activation at low temperature [37]. Jiao et al. has demonstrated that the rate controlling step of CH4 dissociation is the adsorption process and the key intermediate is CHO on Ni(111) surface [32]. Baroni et al. has designed a complex of Rh@Cu(111) as model catalyst, which can hinder the second dehydrogenation step (CH3 → CH2 + H) with respect to the first (CH4 → CH3 + H) [33]. Boukelkoul et al. have doped Cu(100) surface with W, which proved to be effective for CH4 dissociation [38]. Gong et al. has studied conversion between CH4 and CH3 on both flat and stepped Co(0001) surface and suggested that the reaction is not sensitive to structure [35]. However, to the best of our knowledge, the insertion of CO2 into the metal-CH3 site is also essential for CH3COOH formation. Wang et al. has suggested that CO2 insertion into Cu-CH3 bond is the most favorable path in the formation of CH3COOH via comprehensive comparison of different pathways [5]. Ge et al. has studied the C-C coupling reaction on a Zn-doped ceria catalyst. The results show that the Zn dopant may stabilize the methyl group and further contribute the formation of CH3COOH from CH4 and CO2 [39]. Park et al. have indicated that the M+ cation in the M+−ZSM-5 catalyst can assist the activation of CO2 and subsequent reaction with CH3 [40]. However, the origin of reactivity still remains elusive. Besides, the mechanism of the interaction between adsorbed molecules and catalyst need to be revealed in more details in current studies.
In our previous study, we have successfully predicted a two dimensional BC3N2 (2D BC3B2) substrate. This structure is like graphene with B, C and N atoms bonded together upon sp2 hybridization. Compared with graphene, boron nitride and other graphene materials, each hexagonal primitive unit cell of the BC3N2 has one more electron than them. Such a multi-electron system may be beneficial to catalysis due to its advantages in electron transfer rate and effective mass, which is beneficial than other B/C substrates like B2C [5, 41-44].
Inspired by the pioneering works, we attempted to design a novel 2D BC3N2 based catalyst with single metals loaded on it (M@2D-BC3N2) and calculated properties via density functional theory. Among these candidates, Pt@2D-BC3N2 is the most promising catalyst to realize effective activation of CH4 and CO2 with ultralow barriers for CH4 splitting (0.26 eV) and CO2~CH3 combination (0.86 eV). The selectivity for CH3COOH is also very high, which mainly stems from the unique electronic properties of molecules and substrate: The delocalized π bonds in CO2, caused by degenerated states of s, px, py and pz between C and O, can interact dramatically with states of Pt(s), Pt(pz), Pt(dxz), Pt(dyz) and Pt(z2) in Pt@2D-BC3N2. The kinetics model also proves that our system can promote CH3COOH production via simply increasing the temperature or the coverage of CH4 and CO2. Our results provide a reasonable illustration in clarifying mechanism and propose promising candidates with high reactivity for further study.
All the first-principles spin-polarized calculations were performed by using the Vienna ab initio simulation package (VASP) [45, 46]. The ion-electron interactions were described by the projector augmented wave method [47]. The generalized gradient approximation (GGA) is described in the Perdew Burke Ernzerhof form and the cutoff energy for plane-wave basis is set as 400 eV. The convergence criterion for the residual force and energy was set to 0.05 eV/Å and 10−5 eV, respectively, during the structure relaxation. And for all calculations, we considered the vander Waals force correction by using MBD approach in the Grimme scheme [48]. Supercells consisting of 3 × 3 × 1 2D BC3N2 unit cells were used and the Brillouin zones were sampled by a Monkhorst-Pack k-point mesh with a 2 × 2 × 1 k-point grid for structure relaxation, while denser k-points mesh of 7 × 7 × 1 were used for electronic property evaluations. The Bader charge analysis was employed for the charge transfer. A vacuum space of 15 Å was employed to avoid the interaction between two periodic units. The above methods have been widely used in pioneering studies especially in catalytic areas [49-81].
Our predicted 2D BC3N2 exhibits a hexagonal network structure (Fig. 1 and Fig. S1 in Supporting information). In 2D BC3N2, each unit cell has one more electron, leading to a high electron-transfer rate (more details can be obtained in Ref [30]). This character may be beneficial to activate inert molecules such as CO2 and CH4. We thus choose 2D BC3N2 as the substrate for single metal atoms to construct a novel catalyst (denoted as M@2D-BC3N2) in production of CH3COOH via CO2 and CH4.
We first discuss the adsorption sites for single metal atoms loaded on 2D BC3N2. To identify the most stable M@2D-BC3N2 configurations, metal atoms is placed on various sites above BC3N2 (Fig. S1). Each situation is fully relaxed and the most stable structures are confirmed based on systems with the largest binding energies (Eb) for metal atoms on 2D BC3N2 as summarized in Table S1 (Supporting information). The binding energies are generally larger than −2 eV except for Ag, indicating the M@2D-BC3N2 complex is stable. In other words, the binding strength between 2D-BC3N2 and metal atoms is big enough to prohibit the aggregation of single atoms into clusters.
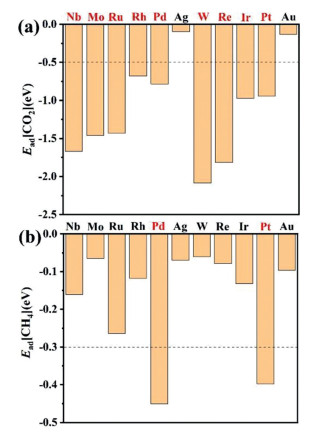
The optimum adsorption configurations of CO2 are discussed comprehensively with different orientations. This is due to the linear character of CO2 molecule, which may lead to various adsorption behavior on single-atom sites (Fig. S2 in Supporting information). After calculation of adsorption energies (Ead), the favorable orientation is assumed as the CO2 molecule adsorbed on metal-atom site with C-O bond pointing to the N atom in the hexagon unit. About the coordination number, CO2 tends to binds M@2D-BC3N2 bidentately with both C and O atoms. Similar conclusions can be obtained in other M@2D-BC3N2 systems. The final structures after fully geometric optimization are displayed in Fig. S3 (Supporting information).
As shown in Fig. 2, the adsorption energies for CO2 are generally larger than −0.5 eV except for Ag and Au, which is remarkably larger than that of CH4 (Ead[CH4] are generally lower than −0.3 eV). Therefore, the adsorption process of CH4 is treated as the rate determined step for simultaneous activation of CO2 and CH4. Pt@2D-BC3N2 and Pd@2D-BC3N2 possess the largest adsorption energy of −0.40 eV and −0.45 eV. However, Pt possesses significant advantages in binding energy on 2D BC3N2 than Pd (Eb[Pt] = −3.91 eV vs. Eb[Pd] = −2.15 eV). We thus serve Pt@2D-BC3N2 system as the most promising candidate for CH3COOH production.
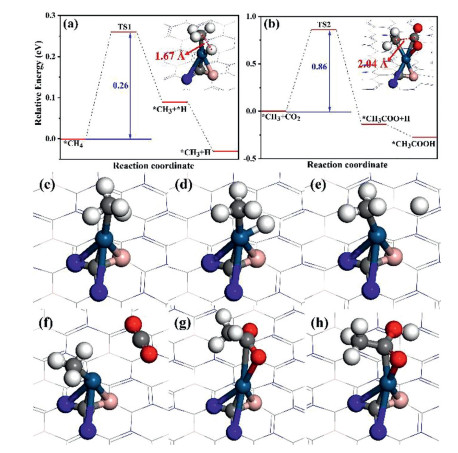
In order to determine the accurate reactivity of the reaction, we have performed many analysis such as reaction energy, transition states and frequency. The reaction begins with the adsorption of CH4 on Pt@2D-BC3N2 and further dissociates to *CH3 and *H. Additional combination between CO2 and *CH3 can form *CH3CO2 intermediate, which can eventually be hydrogenated into CH3COOH as final product. For the co-adsorption state of *CH3 and *H, the *H can spontaneously separate away from *CH3 with a negative reaction energy of −0.12 eV. In contrast, CO2 cannot adsorb together with CH3 on Pt@2D-BC3B2 and combine directly with *CH3 in gas phase to produce CH3COO. We first consider the CH4 → *CH3 + *H step: The catalytic reactivity of CH4 dehydrogenation is surprisingly high with an ultralow reaction barrier of 0.26 eV (Fig. 3), validating the ability of Pt@2D-BC3N2 in activating CH4. The length of C-H bond in CH4 is 1.10 Å at initial state and has enlarged into 2.15 Å after dissociation. The configuration of transition state is shown in the inset of Fig. 3 with a C-H bond length of 1.67 Å. The rate determined step (RDS) of CH4 + CO2 → CH3COOH reaction is *CH3 + *H + CO2 → CH3CO2 + *H step with a reaction barrer of 0.86 eV, which can be easily overcome at only 70 ℃. The CH3CO2 product possesses a C-C bond length of 1.52 Å, which is 2.04 Å in transition states as shown in Fig. 3 and Table S2 (Supporting information). The summarized reaction processes for steps of CH4 → *CH3 + *H and *CH3 + *H + CO2 → CH3CO2 + *H are demonstrated in Fig. S4 (Supporting information) and the continuous process for CH3COOH production is shown in Fig. S5. The only one imaginary frequency of −552.11 cm−1 for CH3~H (−419.23 cm−1 for CH3~CO2) indicates the transition state has been searched correctly for these steps (Table S3 in Supporting information). The results calculated above has illustrated that Pt@2D-BC3N2 could be a dramatically efficient catalyst for activation of inert gasses into CH3COOH.
In addition to CH3COOH, HCOOH is also an important production that we cannot ignore. The major intermediates for HCOOH production include *COOH and *HCOO, which derived from the combination between *H and *CO2. We thus calculate the reaction barriers of *CO2 + *H → *COOH and *CO2 + *H → *HCOO. The reaction barriers are 1.51 eV and 1.01 eV for *COOH and *HCOO pathways (Figs. S6 and S7 in Supporting information), which are all larger than CH3COOH production (0.86 eV). This may indicate that HCOOH production may be hindered by *CO2 hydrodyzation process, leading to the higher selectivity for CH3COOH comapred to HCOOH. We therefore focuse our study on the CH3COOH production on Pt@2D-BC3N2.
Our designed Pt@2D-BC3N2 catayst possesses both notably high adsorption ability and low barrier compared to TM systems. Especially for CH4 dissociation, this is a general rate controlling step on transition metals (TMs) in pioneering studies as shown in Table 1. The high barriers on TMs (0.92~1.78 eV) generally resulted from the weak binding strength (−0.02~−0.04 eV) as the situations on Co, Ni, and Cu. In comparison, Pt exhibits higher CH4 adsorption energy of −0.25 eV, leading to a reduced activation barrier of 0.55 eV. Therefore, the effective capture ability of CH4 can be treated as the origin for high reactivity of CH4 splitting. The C-H bond has been enlonged to 1.67 Å, which is much longer than that on transition metals (1.44~1.55 Å). This may illustrate that CH4 has been sufficiently activated on Pt@2D-BC3N2. Thus, the interaction of CH4 with the substrate can weaken the strength of C-H bond and eventually decrease the dissociation barrier of CH4.
 DownLoad:
CSV
DownLoad:
CSV

|
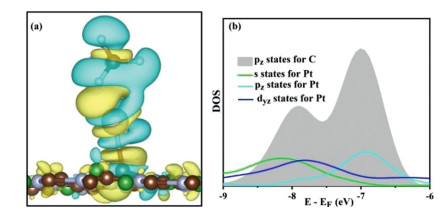
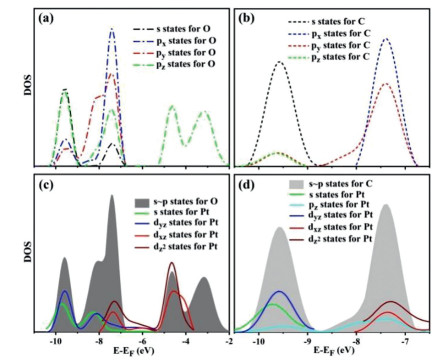
To clarify the ultrahigh reactivity of Pt@2D-BC3N2 in CH3COOH production, we focus our research on the activation of CO2 and CH4 in this part mainly from the hybridizations between various states. For CH4 adsorption, the charge density difference between CH4 and Pt@2D-BC3N2 demonstrates the dramatic accumulation of electrons between Pt atom and C atom in CH4, indicating a covalent bond nature (Fig. 4). The density of states (DOS) of Pt and C atoms can reveal the interaction as hybridization between C(pz), Pt(s), Pt(Pz) and Pt(dyz) states.
For CO2 adsorption, the interaction of CO2 molecules with active sites leads to changes in the geometrical properties of CO2: The angle of O-C-O bond has changed to 117.22° after adsorption, which is in contrast with the value of 180° in free state for CO2. This may indicate the initial steps of chemical adsorption. As mentioned above, CO2 tends to bond with Pt atom with both C and O atoms, which possess strongly degenerate between states, including s, px, py, and pz. This may indicate the existence of delocalized π bonds in CO2 as shown in Figs. 5a and b. The degenerate s~p states of O and C will further interact with Pt states, such as s, pz, dyz, dxz and dz2 (Figs. 5c and d). The overlapping of electronic density regions between Pt site and CO2 may lead to charge transfer between substrate and adsorbed species (Fig. S9 in Supporting information). The bader charge analysis has shown that Pt@2D-BC3N2 has transferred −0.37 e charge to CO2, leading to a colvalent bond interaction between CO2 and Pt@2D-BC3N2. For the CO2 adsorbed on Pt@2D-BC3N2, the valence state of C (O) atom is about +1.63 (−1) (Fig. S8 in Supporting information). The valence is quantified using bader charge with CO2 negatively charged with −0.37 e, which is agreed with the situation of Cu. The electrons can move to the lowest unoccupied orbitals of adsorbed CO2 and eventually stabilize the systems. The characters described above has comprehensively illustrated that CO2 has been totally activated on Pt@2D-BC3N2. Above all, the high reactivity of Pt@2D-BC3N2 mainly stems from the high capture ability of CO2 and CH4 molecules. Moreover, Pt is a material with high cost, which may hinder the practical application. Thus, more research works should be done in our further studies to find out proper substitutes based on the theories proposed in this work.
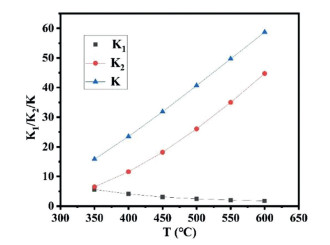
For quantitative analysis, we have calculated the rate constant of CH4 dissociation and CO2~CH3 combination and listed in Table 2. As mentioned above, the barrier of RDS is 0.86 eV, indicating a temperature of 344 K is at least necessary to activate the reaction. We thus investigated the rate constant (k) of CO2 + *CH3 → *CH3COO and its related step of CH4 disscociation at a temperature range of 350~600 K.
DownLoad:
CSV

|
The relation between the reaction rate (r) and corresponding k can be described as follows:
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
|
|
(6) |
|
|
(7) |
|
|
(8) |
where rf (rb) is the reaction rate of forward (backward) reaction; k is the rate constant; θ is the coverage of the adsorbate; A (B, C) is the frequency factor of the reactant (transition state, product); fr (fTS, fp) is the frequency of the reactant (transition state, product); K is the equilibrium constant; Eb is the reaction barrier of RCS; NA is the Avogadro constant. The reaction can reach the equilibrium state at the condition of rf = rb.
From the formulas above, we can deduce the relationship between K and coverage of CH4, CO2 and CH3COO:
|
|
where K1 (K2) is the equilibrium constant of CH4 dissociation (CO2~CH3 combination); K is defined as 
From the data listed in Table 2, we can find that the K1 (K2) is (decreased) increased with increasing temperature due to the endothermic (exothermic) reaction character. However, K constantly exhibits increased tendency with increasing temperature. Based on the formula, the production of CH3COO can be improved via increasing the temperature or the coverage of CH4 and CO2. This can be attributed to the neglegible change tendency between K1 and T compared to K2 situation as shown in Fig. 6. Therefore, K~T exhibits similar tendency as K2~T.
In conclusion, we have performed density functional theory calculation to study M@2D-BC3N2 systems for CH3COOH synthesis via CO2 and CH4. We find that Pt@2D-BC3N2 can dissociate CH4 (aggregate CO2 and CH3) with an ultralow barrier of 0.26 eV (0.86 eV). The high reactivity and selectivity originate from the high capture ability for CO2 and CH4. This phenomenon can be interpreted from the hybridization between various states: The C(pz) states for CH4 can interact with states of Pt(s), Pt(pz) and Pt(dyz) for Pt@2D-BC3N2 complex. Besides, the degenerate between states of s, px, py and pz in C and O in CO2 has indicated the delocalized π bonds. This degenerate states can interact with states of Pt(s), Pt(pz), Pt(dxz), Pt(dyz) and Pt(z2) for Pt@2D-BC3N2. From kinetics aspect, our system can promote CH3COOH production via simply increasing the temperature or the coverage of CH4 and CO2. Our study has not only provided clues for catalyst design in CH3COOH synthesis via CO2 and CH4, but also enriched the understanding of mechanism in the activation of CO2 and CH4.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This study was funded by the National Natural Science Foundation of China (No. 21603109), the Henan Joint Fund of the National Natural Science Foundation of China (No. U1404216), the Special Fund of Tianshui Normal University, China (No. CXJ2020-08), the Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JK0676). We also appreciate the National Supercomputing Center in Zhengzhou to provide the computational sources
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.018.
W. Wang, S.P. Wang, X.B. Ma, J.L. Gong, Chem. Soc. Rev. 40 (2011) 3703–3727. doi: 10.1039/c1cs15008a
M. Aresta, A. Dibenedetto, E. Quaranta, J. Catal. 343 (2016) 2–45. doi: 10.1016/j.jcat.2016.04.003
C. He, R. Wang, D. Xiang, et al., Appl. Surf. Sci. 509 (2020) 145392. doi: 10.1016/j.apsusc.2020.145392
E. McFarland, Science 338 (2012) 340–342. doi: 10.1126/science.1226840
R. Zhang, L. Song, H. Liu, B. Wang, Appl. Catal. A 443 (2012) 50–58.
P. Tang, Q. Zhu, Z. Wu, D. Ma, Energy Environ. Sci. 7 (2014) 2580–2591. doi: 10.1039/C4EE00604F
W. Huang, K.C. Xie, J.P. Wang, et al., J. Catal. 201 (2001) 100–104. doi: 10.1006/jcat.2001.3223
E.M. Wilcox, G.W. Roberts, J.J. Spivey, Catal. Today 88 (2003) 83–90. doi: 10.1016/j.cattod.2003.08.007
J.J. Spivey, E.M. Wilcox, G.W. Roberts, Catal. Commun. 9 (2008) 685–689. doi: 10.1016/j.catcom.2007.08.023
X. Xie, T. Otremba, P. Littlewood, et al., ACS Catal. 3 (2013) 224–229. doi: 10.1021/cs3003963
L.L. Xu, H.L. Song, L.J. Chou, ACS Catal. 2 (2012) 1331–1342. doi: 10.1021/cs3001072
M.M. Nair, S. Kaliaguine, F. Kleitz, ACS Catal. 4 (2014) 3837–3846. doi: 10.1021/cs500918c
A.G. Bhavani, W.Y. Kim, J.S. Lee, ACS Catal. 3 (2013) 1537–1544. doi: 10.1021/cs400245m
F. Polo-Garzon, D. Pakhare, J.J. Spivey, D.A. Bruce, ACS Catal. 6 (2016) 3826–3833. doi: 10.1021/acscatal.6b00666
K. Yuan, J. -. Q. Zhong, X. Zhou, et al., ACS Catal. 6 (2016) 4330–4339. doi: 10.1021/acscatal.6b00357
L. Lin, Z. Shi, J. Huang, et al., Appl. Surf. Sci. 514 (2020) 145900. doi: 10.1016/j.apsusc.2020.145900
L. Lin, J. Huang, W. Yu, et al., Commun. Theor. Phys. 72 (2020) 035501. doi: 10.1088/1572-9494/ab690d
L. Fu, R. Wang, C. Zhao, et al., Chem. Eng. J. 414 (2021) 128857. doi: 10.1016/j.cej.2021.128857
X. Fu, H. Yang, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 1089–1094. doi: 10.1016/j.cclet.2020.08.031
N. Shahzad, B. Khan, J. Mol. Graph. Model. 105 (2021) 107896. doi: 10.1016/j.jmgm.2021.107896
Y. Zhao, H. Wang, J. Han, et al., ACS Catal. 9 (2019) 3187–3197. doi: 10.1021/acscatal.9b00291
W. Huang, W.Z. Sun, F. Li, Aiche J. 56 (2010) 1279–1284.
J. Li, L. Dou, Y. Gao, et al., J. CO2 Util. 52 (2021) 101675.
M. Dutra, M. Schmal, R. Guardani, Caltal. Lett. 148 (2018) 3413–3430. doi: 10.1007/s10562-018-2538-6
G. Wen, Q. Wang, R. Zhang, et al., Phys. Chem. Chem. Phys. 18 (2016) 27272–27283. doi: 10.1039/C6CP05139A
H. Huang, Y. Yu, M. Zhang, Phys. Chem. Chem. Phys. 22 (2020) 27320–27331. doi: 10.1039/c9cp07003f
K. Ray, A.S. Sandupatla, G. Deo, Int. J Quantum Chem. 121 (2021) e26580.
J.M.H. Lo, T. Ziegler, J. Phys. Chem. C 112 (2008) 13642–13649. doi: 10.1021/jp8038219
W. An, X.C. Zeng, C.H. Turner, J. Chem. Phys. 131 (2009) 174702. doi: 10.1063/1.3254383
C.J. Zhang, P. Hu, J. Chem. Phys. 116 (2002) 322–327. doi: 10.1063/1.1423663
A.T. Anghel, D.J. Wales, S.J. Jenkins, D.A. King, Phys. Rev. B 71 (2005) 113410. doi: 10.1103/PhysRevB.71.113410
S.G. Wang, X.Y. Liao, J. Hu, et al., Surf. Sci. 601 (2007) 1271–1284. doi: 10.1016/j.susc.2006.12.059
A. Kokalj, N. Bonini, S. de Gironcoli, et al., J. Am. Chem. Soc. 128 (2006) 12448–12454. doi: 10.1021/ja060114w
J.M. Wei, E. Iglesia, J. Catal. 225 (2004) 116–127. doi: 10.1016/j.jcat.2003.09.030
X.Q. Gong, R. Raval, P. Hu, J. Chem. Phys. 122 (2005) 024711. doi: 10.1063/1.1829257
G.T. de Jong, D.P. Geerke, A. Diefenbach, F.M. Bickelhaupt, Chem. Phys. 313 (2005) 261–270. doi: 10.1016/j.chemphys.2005.01.017
M. Kim, A.D. Franklin, R. Martin, et al., J. Catal. 383 (2020) 181–192. doi: 10.1016/j.jcat.2020.01.027
H. Khettal, M.F. Haroun, M. Boukelkoul, Comput. Theor. Chem. 1186 (2020) 112890. doi: 10.1016/j.comptc.2020.112890
Y. Zhao, C. Cui, J. Han, et al., J. Am. Chem. Soc. 138 (2016) 10191–10198. doi: 10.1021/jacs.6b04446
A.M. Rabie, M.A. Betiha, S. -. E. Park, Appl. Catal. B 215 (2017) 50–59. doi: 10.1016/j.apcatb.2017.05.053
C. He, R. Wang, H. Yang, et al., Appl. Surf. Sci. 507 (2020) 145076. doi: 10.1016/j.apsusc.2019.145076
J. Yu, C. He, C. Pu, et al., Chin. Chem. Lett. 32 (2021) 3149–3154. doi: 10.1016/j.cclet.2021.02.046
H. Yang, C. He, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3202–3206. doi: 10.1016/j.cclet.2021.03.038
R. Wang, C. He, W. Chen, et al., Chin. Chem. Lett. 32 (2021) 3821–3824. doi: 10.1016/j.cclet.2021.05.024
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758–1775.
Furthmuller Kresse, Phys. Rev. B 54 (1996) 11169–11186. doi: 10.1103/PhysRevB.54.11169
Blochl, Phys. Rev. B 50 (1994) 17953–17979. doi: 10.1103/PhysRevB.50.17953
L.M. Azofra, D.R. MacFarlane, C.H. Sun, Phys. Chem. Chem. Phys. 18 (2016) 18507–18514. doi: 10.1039/C6CP02453J
J.R. Huo, L. Fu, C.X. Zhao, C.Z. He, Chin. Chem. Lett. 32 (2021) 2269–2273. doi: 10.1016/j.cclet.2020.12.059
S. Jian, Z. Tian, J. Hu, et al., Adv. Powder Mater. 1 (2022) 100004. doi: 10.1016/j.apmate.2021.09.004
D.Y. Li, L.L. Liao, H.Q. Zhou, et al., Mater. Today Phys. 16 (2021) 100314. doi: 10.1016/j.mtphys.2020.100314
L. Fu, L. Yan, L. Lin, et al., J. Alloys Compd. 875 (2021) 159907. doi: 10.1016/j.jallcom.2021.159907
X. Chen, W.J. Ong, X. Zhao, et al., J. Energy Chem. 58 (2021) 577–585. doi: 10.1016/j.jechem.2020.10.043
J.Y. Luo, P.P. Han, Z.H. Dan, et al. Rare Metals 40 (2021) 3531–3542. doi: 10.1007/s12598-020-01700-1
B. Jackson, J. Chem. Phys. 153 (2020) 034704. doi: 10.1063/5.0012252
A. Gutierrez-Gonzalez, M.E. Torio, H.F. Busnengo, R.D. Beck, Top. Catal. 62 (2019) 859–873. doi: 10.1007/s11244-019-01170-5
Y. Jia, F. Li, K. Fan, L. Sun, Adv. Powder Mater. 1 (2022) 100012. doi: 10.1016/j.apmate.2021.10.003
W. Song, J. Wang, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3137–3142. doi: 10.1016/j.cclet.2021.02.043
M. Mohebinia, C. Wu, G. Yang, et al., Mater. Today Phys. 16 (2021) 100293. doi: 10.1016/j.mtphys.2020.100293
X. Chen, W.J. Ong, Z. Kong, et al., Sci. Bull. 65 (2020) 45–54. doi: 10.1016/j.scib.2019.10.016
Y. Sun, Z. Deng, X. -. M. Song, et al., Nano-Micro Lett. 12 (2020) 133. doi: 10.1007/s40820-020-00444-y
Y. Wang, M. Batmunkh, H. Mao, et al., Chin. Chem. Lett. 33 (2022) 394–398. doi: 10.31083/j.rcm2312394
X. Lv, W. Wei, F. Li, et al., Nano Lett. 19 (2019) 6391–6399. doi: 10.1021/acs.nanolett.9b02572
H. Hu, H. Yang, X. Yang, et al., Chin. Chem. Lett. 31 (2020) 3213–3215. doi: 10.1016/j.cclet.2020.08.025
Z. Li, Z. Ma, J. Liang, et al., Mater. Today Phys. 22 (2022) 100586. doi: 10.1016/j.mtphys.2021.100586
G. Zhao, W. Ma, X. Wang, et al., Adv. Powder Mater. 1 (2022) 100008. doi: 10.1016/j.apmate.2021.09.008
X. Lv, W. Wei, B. Huang, et al., Nano Lett. 21 (2021) 1871–1878. doi: 10.1021/acs.nanolett.0c05080
H. Jing, P. Zhu, X. Zheng, et al., Adv. Powder Mater. 1 (2022) 100013. doi: 10.1016/j.apmate.2021.10.004
X.J. Fang, L.P. Ren, F. Li, Z.X. Jiang, Z.G. Wang, Rere Metals 41 (2022) 901–910. doi: 10.1007/s12598-021-01819-9
Z.Q. Gao, C.Y. Wang, J.J. Li, et al., Acta Phys. 37 (2021) 2010025.
T.T. Wu, W.J. Fan, Y. Zhang, F.X. Zhang, Mater. Today Phys. 16 (2021) 100310. doi: 10.1016/j.mtphys.2020.100310
F. Rao, G. Zhu, W. Zhang, et al., ACS Catal. 11 (2021) 7735–7749. doi: 10.1021/acscatal.1c01251
F. Rao, G.Q. Zhu, W.B. Zhang, et al., Appl. Catal. B 281 (2021) 119481. doi: 10.1016/j.apcatb.2020.119481
Y.H. Wang, R.Q. Li, H.B. Li, et al., Rare Metal. 40 (2021) 1410–1417.
L. Yang, Z. Liu, S. Zhu, et al., Mater. Today Phys. 16 (2021) 100292. doi: 10.1016/j.mtphys.2020.100292
C. Pu, J. Yu, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 1081–1085. doi: 10.1016/j.cclet.2020.08.042
M. Jin, W.H. Yang, X.H. Wang, et al., Rare Metals 40 (2021) 858–864. doi: 10.1007/s12598-020-01601-3
S. Li, Y. Wang, J. Liang, et al., Mater. Today Phys. 18 (2021) 100396. doi: 10.1016/j.mtphys.2021.100396
T. Zhang, C.T. Liu, Adv. Powder Mater. 1 (2022) 100014. doi: 10.1016/j.apmate.2021.11.001
Y.Z. Wang, Y.M. Ding, C.H. Zhang, et al., Rare Metals 40 (2021) 2785–2792. doi: 10.1007/s12598-021-01765-6
D. Zhou, C. Li, F. Yin, et al., Chin. Chem. Lett. 31 (2020) 2325–2329. doi: 10.1016/j.cclet.2020.04.045

Figure 1 (a) The configuration of 2D BC3N2 with metal atom loaded on it. The atom in the red cycle denotes the doped metal atom. (b) Binding energies of metal atoms on 2D BC3N2.

Figure 3 The reaction pathways of (a) CH4 dissociation and (b) combination of CO2 and CH3. The configurations of transition states are displayed in the insets of (a) and (b). The configurations of (c) *CH4, (d) *CH3 + *H, (e) *CH3 + H, (f) *CH3 + CO2, (g) *CH3COO, and (h) CH3COOH on Pt@2D-BC3N2.

Figure 4 (a) The charge density difference and (b) density of states (DOS) for hybridization between Pt and C atoms for CH4 adsorbed on Pt@2D-BC3N2. The charge depletion and accumulation were depicted by cyan and yellow, respectively.

Figure 5 Density of states (DOS) of degenerate between s and p states in (a) O and (b) C. The DOS in (c) and (d) also exhibit the degenerate states of (c) O and (d) C with s, p and d states in Pt.
Table 1. Various reactivity data of CH4 dissociation including activation barriers (Ea), bond strength of C-H for transition states (d(C-H)), and adsorption energies of CH4 (Ead[CH4]) for systems of Co, Ni, Cu, Pt, Ir, and our designed Pt@2D-BC3N2.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

