Citation:
Wei Li, Hechen Wu, Xunlei Ding, Xiaonan Wu. The cycloaddition reaction of ethylene and methane mediated by Ir+ to generate a half-sandwich structure IrHCp+[J]. Chinese Chemical Letters,
2023, 34(1): 107196.
doi:
10.1016/j.cclet.2022.02.002
The cycloaddition reaction of ethylene and methane mediated by Ir+ to generate a half-sandwich structure IrHCp+
English
The cycloaddition reaction of ethylene and methane mediated by Ir+ to generate a half-sandwich structure IrHCp+
Received Date:
24 December 2021 Accepted Date:
01 February 2022 Revised Date:
19 January 2022 Available Online:
15 January 2023
Abstract:
The cycloaddition reactions of methane and ethylene mediated by Ir+ have been designed and studied by the techniques of mass spectrometry in conjunction with theoretical calculations. Studies have shown that Ir+ can mediate the cycloaddition reaction of CH4 and two C2H4 to generate a half-sandwich structure IrHCp+ (Cp = η5-C5H5) including pentamethylcyclopentadienyl ligand by continuous dehydrogenation reaction with the forming of three C-C bonds and seven C-H bonds. The orbital analysis indicates the mechanism of the cyclization reaction to generation of pentamethylcyclopentadienyl ligand with odd number carbon atom depends on the overlap of π orbitals in -C2H2 and carbene, which is more difficult than the forming of cyclobutadiene ligand and benzene. This study may help to understand the reaction mechanism in the cycloaddition reactions of organic compounds, which will be useful to guide the rational design of new catalysts with tailored selectivity and increased efficiency.
The cycloaddition reactions of small organic molecules involving alkane, alkene and alkyne have been a topic for intense investigation during the past decades as one of the most straightforward methods to produce useful carbocyclic systems [1-4]. Due to the thermodynamic stability and kinetic inertness of small organic molecules, the cycloaddition reactions for them are difficult to occur, which always need extremely high temperature or high pressure [5-8]. Therefore, the catalyst is essential for the cycloaddition reactions. Transition metal compounds are effective in catalyzing the cycloaddition reactions by changing its spin state due to the empty or half-filled d orbitals of transition metals, which have become the most abundant industrial catalysts for cycloaddition reactions [9-13].
In the past decades, the researchers applied different forms of transition metal catalysts including the bare transition metal atoms (Co, Ti, Y, Zr, Nb, Ni, Ru, Rh and Mo, etc.) [14-18], metal oxides (TiO2, VO2) [19-21], neutral metal clusters (Pdn), ionic metal clusters (Fen+) [22] and so forth to study the reaction of acetylene cyclization through experiments and theoretical calculations [23-29]. The intermediate products M(η2−C2H2)+, M(η2-C4H4)+, M(η4-C4H4)+, M(η2-C6H6)+ and M(η6-C6H6)+ are generated. Their structures have been characterized in the gas phase experiments with theoretical calculations. M(η2−C2H2)+, M(η2-C4H4)+ and M((η2-C6H6)+ are metal-ligand ring structures which can be described by the Dewar-Chatt-Duncanson (DCD) complexation model [30-35]. According to this model, the interaction of metals and ligands can be described as σ donation and π back donation [30-35]. M(η4-C4H4)+ and M((η6-C6H6)+ with metal cation-π structure contain cyclobutadiene and benzene, which are well-known in organometallic chemistry, and even been employed in gas-phase ion chemistry [30-35].
In addition to the reactions to generate the cyclobutadiene ligand and benzene with even numbers of carbon atom, the cycloaddition reaction to form pentamethylcyclopentadienyl (Cp) ligand with odd numbers of carbon atom is also important type of reaction in the fields of organic synthesis, medicinal chemistry, pesticides, and chemical industry [36, 37]. As far as we know, the reaction to obtain a ring with odd numbers of carbon atom has not been obtained in the gas phase. Because the ground state of Ir+ is 5d76s1, which has the enough empty valence orbital (of suitable symmetry) for the cycloaddition reaction. The iridium and iridium complexes have been applied to mediate the different cyclization reaction of small organic molecules [38-41]. Therefore, in order to obtain the five-membered pentamethylcyclopentadienyl ring ligand, in this work, the cycloaddition reactions of methane and ethylene mediated by Ir+ have been designed and studied by the techniques of mass spectrometry in conjunction with theoretical calculations. We will clearly investigate the generation of the half-sandwich structure IrHCp+ (Cp = η5-C5H5) including pentamethylcyclopentadienyl ligand by high selectivity, the potential energy surfaces of consecutive reactions in different electronic states and the bond analysis of the products to further understand the special cycloaddition reaction and guide the rational design of new catalysts.
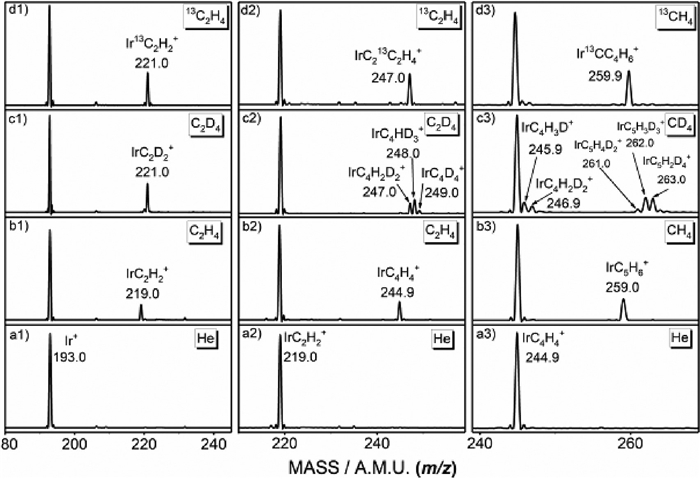
The experiments are performed by an ion trap mass spectrometer equipped with a laser vaporization-supersonic expansion ion source coupled with a flow tube reactor, which has been reported previously [42, 43]. For getting pentamethylcyclopentadienyl ligand, the cycloaddition reaction routes Ir+ → IrC2H2+ → IrC4H4+→ IrC5H6+ are designed. The metal ions Ir+ are generated by pulsed laser ablation of a rotating and translating metal Ir target. The 532 nm second harmonic of a Nd: YAG laser with an energy of 5–12 mJ/pulse is used. The nascent ablated plasma is entrained by the helium carrier gas (99.999%) expanded from a pulsed valve (General Valve, series 9) at a backing pressure of about 0.3–0.5 MPa. The generated ions are guided and mass-selected by the quadrupole, and then sent into a quadrupole linear ion trap. The mass spectra from the reactions of Ir+ ions with He, C2H4, C2D4 and 13C2H4 in the ion trap are shown in Fig. 1 (panels a1, b1, c1, d1). The results indicate that only one product with chemical formula of IrC2H2+ is generated by reaction 1.
(1)
Figure 1
Figure 1.
Mass spectra from the reactions of [Ir]+ and [IrC2H2]+ with (a1, a2) He, (b1, b2) C2H4, (c1, c2) C2D4 and (d1, d2) 13C2H4, as well as the reactions of [IrC4H4]+ with (a3) He, (b3) CH4, (c3) CD4 and (d3) 13CH4, the specific m/z value for each of peaks of mass spectra are given, respectively.
Isotopic-labeling experiments by using the C2D4 and 13C2H4 (panels c1 and d1) confirm this result with the generation of IrC2D2+ and Ir13C2H2+. Similarly, the reaction of mass-selected IrC2H2+ with C2H4 is studied, and the mass spectra from the reactions with He, C2H4, C2D4, and 13C2H4 are shown in Fig. 1 (panels a2, b2, c2, d2). The observation of only product IrC4H4+ suggests that the reaction 2 takes place:
(2)
Isotopic-labeling experiments by using the C2D4 and 13C2H4 (panels c2 and d2) confirm that the generation of the IrC213C2H4+ and IrC4H2D2+/IrC4HD3+/IrC4D4+.
For getting the five-membered ring ligand, the mass spectra from the reactions of IrC4H4+ with He, CH4, CD4, and 13CH4 are studied and shown in Fig. 1 (panels a3, b3, c3, d3). One peak that can be assigned to the product ions with chemical formula of [IrC5H6]+ is observed in the spectra, reaction 3.
(3)
Isotopic-labeling experiments using the 13CH4 sample confirm that the four carbon atoms in the [IrC5H6]+ product ion are originated from the C2H4 reactant and one carbon atom is originated from the CH4 reactant. However, we point out that when Ar is added for cooling before the reaction gasses are inlet, no products are found for the reactions of [IrC4H4]+ with CH4. For the reactions of IrC2H2+ or Ir+ with C2H4, no similar phenomenon is found.
In order to gain insight into the reaction mechanisms, the geometry optimization and frequency calculations for reactants, products, reaction intermediates (IMs) and transition states (TSs) were carried out by using the TPSS method and the Def2TZVP basis sets by Gaussian 09 software package [44-47]. All the stationary point structures were characterized on the Potential Energy Surface (PES) by performing vibrational frequencies analysis, which aimed at identifying the nature of the stationary point (minima or saddle point). In predicting the reaction pathways, the intrinsic reaction coordinate (IRC) [48, 49] calculations were performed to confirm the correctness of the transition states. All energies are reported by zero-point vibrational energy (ZPE) correction. The analyses of the quantum theory of atoms in molecules (QTAIM) [50, 51], charge decomposition analysis (CAD) [52] and orbital interaction diagram are generated using the Multiwfn package [53].
The various possible structures of dehydrogenation products IrC2H2+, IrC4H4+ and IrC5H6+ are obtained by calculations at the TPSS/def2-TZVP level and shown in Figs. S1-S3 (Supporting information). For IrC2H2+, the most stable structure is metal cation-π complex 3Ir(η2-C2H2)+. The corresponding singlet state structure is predicted to lie 0.09 eV higher in energy than the triplet state. The most stable structure of IrC4H4+ is metallacycle structure 1Ir(η2-C4H4)+ of coupling by -C2H2 (from IrC2H2+) and -C2H2 (activated products of ethylene) which has the ground state 1A1. The 1Ir(η4-C4H4)+ and corresponding triplet state structure 3Ir(η2-C4H4)+ are predicted to lie 0.35 eV and 0.58 eV higher in energy than the most stable structure. The most stable structure of IrC5H6+ is 1IrH(η5-C5H5)+, which is half-sandwich structure with IrH moiety as the center and coordinated with Cp ligand that is obtained from the coupling by -C2H2, -C2H2 and -CH (activated products of methane). The isomers 1Ir(η5-C5H6)+ and 1IrH(η2-C5H5)+ are predicted to lie 0.43 eV and 0.45 eV higher in energy than the most stable structure.
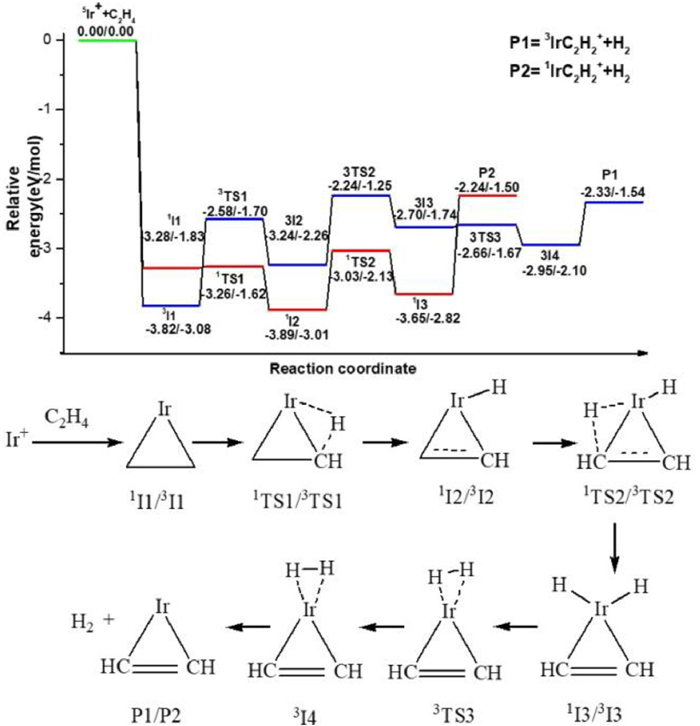
The pathway for the first dehydrogenation reaction Ir+ + C2H4 starting with the quintet state as ground reactants is shown in Fig. 2 and the details are shown in Fig. S4 (Supporting information). An ethene adduct, 5I1 is formed upon initial approach. The efficient dehydrogenation at the thermal energies cannot occur along the quintet state surface because the transition state 5TS2 lies 0.68 eV above the reactants, so further calculations along the quintet state surface are not conducted. A surface crossing must occur from the quintet state to the singlet state or triplet state surface for dehydrogenation. Along the triplet state surface, the ethene adduct, 3I1, lies 3.82 eV lower in energy than the ground state reactants. From 3I1, the first oxidative addition process with the transfer of the first H atom from ethene to the Ir+ occurs through 3TS1 (−2.58 eV) resulting in the formation of the intermediate 3I2 (−3.24 eV). The second oxidative addition process, with C–H bond cleavage from -C2H to the Ir+ center leads to generate 3I3 (−2.70 eV) by a transition state 3TS2 (−2.24 eV). Then, intermediate 3I4 (−2.95 eV) in which H2 is adsorbed by Ir+ through weak interactions is formed with the decrease of H-H distance. The final triplet state product 3Ir(η2-C2H2)+ with metal cation-π structure is generated with the reductive elimination process of H2. Along the singlet state surface, similar to the triplet state surface, this crossing takes place in the entrance channel because intermediate 1I1 lies 3.28 eV below 5I1. Then the reaction occurs through the oxidative addition and reductive elimination processes of C-H bonds. It requires 0.09 eV more energy than the triplet state dehydrogenation process, but still lies 2.24 eV below ground state reactants. Therefore, an effective dehydrogenation reaction can not only occur along the triplet state surface but also along the singlet state surface, the singlet and triplet state products 1Ir(η2-C2H2)+ and 3Ir(η2-C2H2)+ may be coexisting.
Figure 2
Figure 2.
The dehydrogenation pathways for the cycloaddition reaction of Ir+ + C2H4. Relative energies of the reaction intermediates, transition states, and products with respect to the separated ground state reactants are given by TPSS/CCSD(T) (the unit is eV).
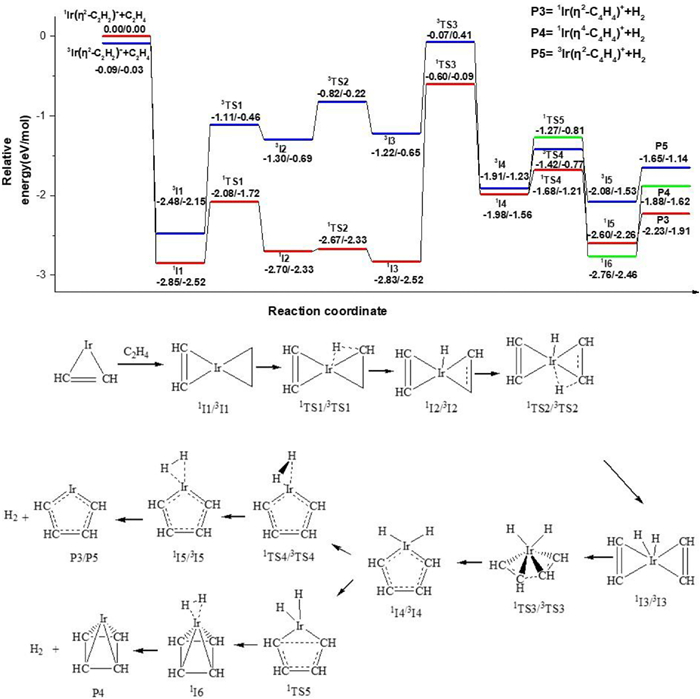
The pathway for the second dehydrogenation reaction, which begins with the singlet state or triplet state Ir(η2-C2H2)+ + C2H4 is shown in Fig. 3 and the details are shown in Fig. S5 (Supporting information). The single state 1Ir(η2-C2H2)+ + C2H4 is determined as ground reactants for discussion. Along the singlet state surface, the adsorption complex, 1I1 lies 2.85 eV lower in energy than the ground state reactants. The first oxidative addition process with the transfer of the first H atom from ethene to the Ir+ occurs through 1TS1 (−2.08 eV) resulting in the formation of the intermediate 1I2 (−2.70 eV). The second oxidative addition process, with C–H bond cleavage from -C2H to the Ir+ center leads to intermediate 1I3 (−2.83 eV) by a transition state 1TS2 (−2.67 eV). The cycloaddition reaction with the coupling of -C2H2 from reactant Ir(η2-C2H2)+ and -C2H2 from the activated product of reactant C2H4 forms a metallacycle structure 1I4 (−1.98 eV) through a transition state 1TS3 (−0.60 eV). Then, intermediate 1I5 (−2.60 eV) in which H2 is adsorbed by the metal cation Ir+ through weak interactions is formed with the decrease of H-H distance. The final metallacycle structure 1Ir(η2-C4H4)+ with singlet state is generated with the reductive elimination process of H2. In addition, the 1Ir((η4-C4H4)+ is also generated through the potential energy surface (1I4–1TS5–1I6). The IRC calculations of the pathways 1I3–1TS3–1I4 and 1I4–1TS5–1I6 are performed and the results are shown in Figs. S6 and S7 (Supporting information). The reaction pathway to generate 3Ir(η2-C4H4)+ for the triplet surface is similar to that of the singlet state. It requires 0.58 eV more energy than the singlet state dehydrogenation process, but still lies 1.65 eV below ground state reactants, so the reaction can proceed along the singlet state and the triplet state surface, the 1Ir(η4-C4H4)+, triplet and singlet products Ir(η2-C4H4)+ are generated. But the energy of 3TS4, 1TS5 is higher than that of 1TS4, and the energy of P4, P5 is higher than that of P3, we predict the main product of IrC4H4+ is 1Ir(η2-C4H4)+.
Figure 3
Figure 3.
The dehydrogenation pathways for the cycloaddition reaction of Ir(η2-C2H2)+ + C2H4. Relative energies of the reaction intermediates, transition states, and products with respect to the separated ground state reactants are given by TPSS/CCSD(T) (the unit is eV).
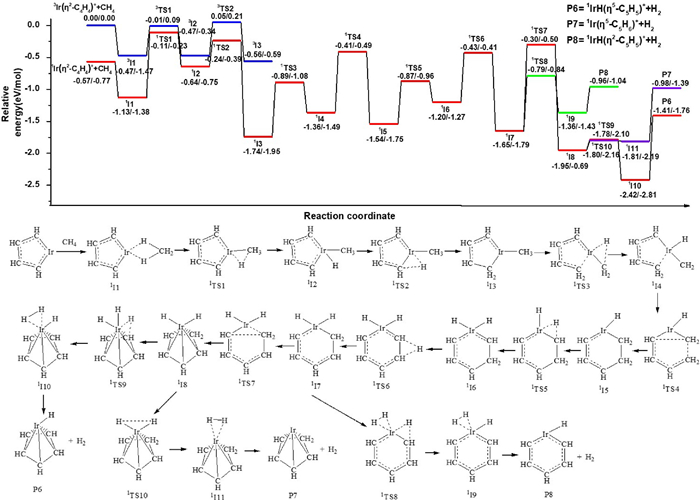
The pathway for the third dehydrogenation reaction is shown in Fig. 4 and the details are shown in Figs. S8 and S9 (Supporting information). The reaction leading to the IrC5H6+ + H2 products is predicted to proceed via oxidative addition, reductive elimination, ring-forming and dehydrogenation processes. If the reaction begins with the singlet state 1Ir(η2-C4H4)+ + CH4 or 1Ir(η4-C4H4)+ + CH4 as the ground reactants, the efficient dehydrogenation at the thermal energies cannot occur because the energy of transition state is higher than the reactants (Figs. S6 and S7), this is consistent of experimental results that no product is observed when ions are cooled by Ar gas. The experimental results can be explained that the part of the release energy in the reactions of IrC2H2+ or Ir+ with C2H4 is not transferred away and the product IrC4H4+ owns the extra energy to overcome the barrier. So the reaction can begin with the triplet state 3Ir(η2-C4H4)+ + CH4 as ground reactants or singlet state with the extra energy [54]. Along the triplet state surface, the ethene adsorption complex, 3I1 lies 0.47 eV lower in energy than the ground state reactants. When the activation of the first C-H bond of CH4 occurs, the second transfer of hydrogen atom leads to the intermediate 3I3 by the transition state 3TS2 which lies 0.05 eV above the reactants. Because of this, efficient dehydrogenation at thermal energies cannot occur along the triplet state surface, so further calculations along the triplet state surface are not conducted. The triplet state Ir(η2-C4H4)+ is directly coordinated to CH4 for forming the ethene adduct, 1I1, lies 1.13 eV lower in energy than the ground state reactants. From 1I1, the first oxidative addition process with the transfer of the first H atom from CH4 to the Ir+ occurs through 1TS1 (−0.11 eV) resulting in the formation of the intermediate 1I2 (−0.64 eV). The first reductive elimination process, with the transfer of H atom from Ir+ to adjacent C atom leads to intermediate 1I3 (−1.74 eV) by a transition state 1TS2 (−0.24 eV). The second oxidative addition process with the transfer of the H atom from -CH3 to the Ir+ occurs through 1TS3 (−0.89 eV) resulting in the formation of the intermediate 1I4 (−1.36 eV). Then the first ring-forming reaction with the coupling of -C4H5 and -CH2 from the activated product of reactant CH4 forms a metallacycle structure 1I5 (−1.54 eV) through a transition state 1TS4 (−0.41 eV). The third oxidative addition process, with the transfer of H atom from -CH2 (the activated product of reactant CH4) to Ir+ atom leads to intermediate 1I6 (−1.20 eV) by a transition state 1TS5 (−0.87 eV). Then the H atom is transferred between the two C atoms to form intermediate 1I7 (−1.65 eV) by a transition state 1TS6 (−0.43 eV). The second ring-forming reaction with the coupling of two C atoms adjacent to Ir+ through 1TS7 (−0.30 eV) results in the formation of the intermediate 1I8 (−0.43 eV) with a like-half-sandwich structure that IrH2+ connects to the five-membered ring -C5H6. The final oxidative addition process, with the transfer of the H atom from -CH2 to the Ir+ center takes place to form the intermediate 1I9, in which H2 is adsorbed by the metal Ir through weak interactions. Finally, the half-sandwich structure IrHCp+ is generated with the reductive elimination process of H2, which is consistent with the experimental results. The generated pathways of isomers Ir(η5-C5H6)+ and IrH(η2-C5H5)+ are exothermic processes which lie 0.98 eV and 0.96 eV below ground state reactants. But the energies of Ir(η5-C5H6)+ and IrH(η2-C5H5)+ are higher than the IrHCp+, Therefore, the IrHCp+ is major product, Ir (η5-C5H6)+ and IrH (η2-C5H5)+ may be also coexisting, which is similar with the isomers of IrC4H4+. The IRC calculations of the pathways 1I3–1TS3–1I4 and 1I4–1TS5–1I6 are performed and the results are shown in Figs. S10 and S11 (Supporting information).
Figure 4
Figure 4.
The dehydrogenation pathways for the cycloaddition reaction of Ir(η2-C4H4)++CH4. Relative energies of the reaction intermediates, transition states, and products with respect to the separated ground state reactants are given by TPSS/CCSD(T) (the unit is eV).
In addition, the single-point energy of dispersion correction, the entropy and Gibbs free energy are calculated and the comparison diagrams are shown in Figs. S12–14 (Supporting information) and the values are given in Tables S1-S3 (Supporting information). The energy trends of dispersion correction and the entropy are consistent with the calculation of the TPSS function used. The single-point calculations using high-level CCSD(T) methods on the reactants, products, reaction intermediates and transition states are shown in Fig. 2, Fig. 3, Fig. 4. The reaction pathways are consistent with the TPSS function except for the 3TS3 of the cycloaddition reaction of Ir(η2-C2H2)+ with C2H4. The relative energy of transition states 3TS3 is higher 0.41 eV than the ground state reactants, but the energy singlet state 1TS3 is lower than the reactant. So the cycloaddition reaction of Ir(η2-C2H2)+ with C2H4 might have a cross point (CP) in the singlet and triplet potential energy surfaces [55]. The reaction products are same as the results of TPSS function, so the calculation of TPSS function is accurate.
In order to explain the mechanism of the cyclization reaction, we analyze the localized molecular orbital (LMO) of the cyclization process and the results are shown in Fig. S15 (Supporting information). For the cyclization process of reaction 1Ir(η2-C2H2)+ + C2H4, from 1I3 to 1I4 through the transition state 1TS3, with the distance between the carbon atoms from 1Ir(η2-C2H2)+ and C2H4 gradually decreases, the two π orbitals of -C2H2 from Ir(η2-C2H2)2+ approach, and then overlap to form a multi-center bond (1TS3), and further overlap to form a new C-C σ bond, the π orbitals of carbons in ring overlap result in forming the four-membered cyclobutadiene ring ligand C4H4. The LMO of the cyclization process above is the same to the that of the reaction 3Ir(η2-C2H2)+ + C2H4 to generate 3Ir(η4-C4H4)+. For the first ring formation process of reaction 1Ir(η2-C4H4)+ + CH4, the two overlapping π orbitals are from the -IrCH2 with carbene structure and -C2H2 of ring η2-C4H4, which leads to form another new C-C σ bond. Finally, the π orbitals of carbons in ring overlap to form the pentamethylcyclopentadienyl ligand (Cp ring). Therefore, the mechanism of the cyclization reaction depends on the overlap of π orbitals which is consistent with the statement of previous paper [13].
In order to classify bond types, characterize bond nature, and distinguish bond strength of the special product IrH(Cp)+ containing pentamethylcyclopentadienyl ligand. The bond analysis is applied by the topological parameters at the bond critical point (BCP) between two atoms based on the quantum theory of atoms in molecules (QTAIM) theory [56−59]. From Table S4 (Supporting information), for the topological parameters the Ir-C bonds of IrH(Cp)+, ρb and ∇2ρb are positive, Hb is negative, and -Gb/Vb is greater than 0.5 and less than 1, which illustrate that IrH(Cp)+ is formed through the dative bond between IrH+ and Cp ligand [56-59]. For the topological parameters the C-C bond of Cp, ρb is positive, ∇2ρb and Hb are negative, and -Gb/Vb is less than 0.5, which illustrate the C-C bond of Cp is covalent bonds [56-59]. The analyses of orbital interaction diagram and charge decomposition analysis (CDA) [60, 61] are applied to understand how orbitals of fragments are mixed to form the dative bond of IrH(Cp)+ (Figs. S16 and S17 in Supporting information). For IrH(Cp)+, the LUMO is mainly composed of the dxz orbital (44%) of 1Ir+ and the π orbital (41%) of Cp, the all occupied molecular orbitals are formed by the s+dz2 orbital (68%) of IrH+ and the π orbital (20%) of Cp, the s+dz2 orbital (25%) of IrH+ and the π orbital (26%) of Cp, the dxz orbital (34%) of 1Ir+ and the π orbital (47%) of Cp, the dxy orbital (34%) of 1Ir+ and the π orbital (14%) of Cp, the s+dz2 orbital (16%) of IrH+ and the π orbital (64%) of Cp. The calculated data of CDA of IrH(Cp)+ indicate that the dative bond of IrH+ and Cp is formed between the donation from IrH+ to Cp and back-donation from Cp to IrH+ by above mixed orbitals. The numbers of donation electron and back-donation electron are 0.1164 and 0.1361 and the back-donation interaction is greater than donation.
Previous studies have shown that catalysts containing transition metals can effectively promote the reaction of acetylene cyclization to form cyclobutadiene ligand and benzene [23-29]. Cp is important ligand of sandwich structure compound whose importance has been shown in the fields of organic synthesis, catalysis, medicinal chemistry, pesticides, and chemical industry [36, 37]. Normally, due to the thermodynamic stability and kinetic inertness, the cycloaddition reaction of ethylene and methane to form pentamethylcyclopentadienyl ligand with the activation and forming of C-C bond and C-H bond does not happen at room temperature [5-8]. Besides, the ring with odd numbers of carbon atom is more difficult to generate than the ring with even numbers of carbon atom, because -C2H2 has the same π orbitals and is easier to overlap to form the ring with even numbers of carbon atom, while the ring with odd numbers of carbon atom have to overlap with carbene, which is a dehydrogenation product methane with different π orbital than that of the -C2H2. Therefore, despite a lot of work on synthesis of cyclobutadiene ligand and benzene, there is no work on the generation of Cp in the gas phase. In this paper, the cycloaddition reaction of two ethylene and methane by the "catalyst" Ir+ can happen to generate the Cp ring ligand by high selectivity, which proceeds by the activation/formation of three C-C bonds and seven C-H bonds and only one products IrC2H2+, IrC4H4+ and IrC5H6+ are found for Ir+/C2H4, IrC2H2+/C2H4 and IrC4H4+/CH4 reactions in this process. The metal ions Ir+ plays a key role, which decreases the energy for C-H and C-C bond activation and promotes the overlap of different π orbital. In addition, our study shows that the products Ir(η2-C2H2)+, Ir(η2-C4H4)+, Ir(η4-C4H4)+ and IrH(Cp)+ are formed by the dative bonds. These may help to understand the reaction cycloaddition mechanism and guide the rational design of new half-sandwich and sandwich catalysts with tailored selectivity and increased efficiency.
In conclusion, the cycloaddition reactions of methane and ethylene mediated by Ir+ are designed, which have been studied by gas-phase experiments with theoretical calculations. Experimental results found the high selectivity reactions of Ir+/C2H4, IrC2H2+/C2H4 and IrC4H4+/CH4, and only one products IrC2H2+, IrC4H4+ and IrC5H6+ are confirmed, respectively. Calculations have shown that Ir+ can mediate the cycloaddition reaction of CH4 and two C2H4 to generate the half-sandwich structure IrHCp+ containing the pentamethylcyclopentadienyl ligand by continuous dehydrogenation reaction with the forming of three C-C bonds and seven C-H bonds. The orbital analysis indicates the mechanism of the cyclization reaction to generation of pentamethylcyclopentadienyl ligand depends on the overlap of -C2H2 and carbene π orbitals, which is more difficult than the overlap of same -C2H2π orbitals to form cyclobutadiene ligand and benzene. The calculated QTAIM and CDA data of IrH(Cp)+ indicate that the dative bond of IrH+ and Cp is formed between the donation from IrH+ to Cp and back-donation from Cp to IrH+. The back-donation interaction is greater than donation. This study may help to understand the reaction mechanism and metal-mediated ability in cycloaddition reaction of organic compounds, which will be useful to guide the rational design of new catalysts with tailored selectivity and increased efficiency.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The work was supported by Beijing Natural Science Foundation (No. 2214064), the National Natural Science Foundation of China (Nos. 21603037, 21688102, 92161115, 21973016, 91545122), the Fundamental Research Funds for the Central Universities (Nos. JB2015RCY03, JB2019MS052) supported by the fund of North China Electric Power University.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.002.
Figure 1
Mass spectra from the reactions of [Ir]+ and [IrC2H2]+ with (a1, a2) He, (b1, b2) C2H4, (c1, c2) C2D4 and (d1, d2) 13C2H4, as well as the reactions of [IrC4H4]+ with (a3) He, (b3) CH4, (c3) CD4 and (d3) 13CH4, the specific m/z value for each of peaks of mass spectra are given, respectively.
Figure 2
The dehydrogenation pathways for the cycloaddition reaction of Ir+ + C2H4. Relative energies of the reaction intermediates, transition states, and products with respect to the separated ground state reactants are given by TPSS/CCSD(T) (the unit is eV).
Figure 3
The dehydrogenation pathways for the cycloaddition reaction of Ir(η2-C2H2)+ + C2H4. Relative energies of the reaction intermediates, transition states, and products with respect to the separated ground state reactants are given by TPSS/CCSD(T) (the unit is eV).
Figure 4
The dehydrogenation pathways for the cycloaddition reaction of Ir(η2-C4H4)++CH4. Relative energies of the reaction intermediates, transition states, and products with respect to the separated ground state reactants are given by TPSS/CCSD(T) (the unit is eV).

 DownLoad:
DownLoad:

 下载:
下载:





