
Figure 1.
XRD patterns (a), the elemental mapping images (b) and NOx conversion curve during NH3-SCR reaction (c) of the synthesized samples.
Investigation of the promotion effect of Mo doped CuO catalysts for the low-temperature performance of NH3-SCR reaction
Hui Wang , Ting Zhu , Yujie Qiao , Shicheng Dong , Zhenping Qu

The emission of nitrogen oxides (NOx, x = 1, 2) from stationary sources and mobile sources has caused atmospheric pollution problems such as acid rain, photochemical smog, and ozone holes [1]. One typical technique, selective catalytic reduction with ammonia (NH3-SCR), has been widely developed to convert NOx to harmless N2 and H2O. A commercial V2O5-WO3(MoO3)/TiO2 catalyst has been used in the removal of NOx for stationary sources [2, 3]. However, there are still some unavoidable disadvantages, such as the narrow operating temperature window between 300 ℃ and 400 ℃, poor activity at the low temperature, and the biological toxicity of vanadium [4]. Therefore, many researchers focus on the novel, eco-friendly, low-cost and high efficient NH3-SCR catalysts. Several transitional metal oxides catalysts, including CuOx, MnOx, CeO2 and Fe2O3 have been attracted great attention in recent years, which are active for the low-temperature NH3-SCR reactions [5-9]. Among these catalysts systems, CuO based catalysts have been intensively studied for low-temperature de-NOx [10]. However, several problems, including lack of Lewis and Brønsted acidity and poor resistance to H2O and SO2 poison remain to overcome [11, 12]. Several strategies, such as modification by mixing with other metal oxides, doping with other ions [8, 13], and dispersion on a high surface area support [14], have been employed to enhance the de-NOx activity of CuO and other transitional metal oxides catalysts. In our previous study [8], the Fe doped CeO2 catalysts with Fe-O-Ce system were developed and enhanced the adsorption of NH3 and NOx, oxygen vacancies, improving the NH3-SCR activity. MoOx has been well recognized as an excellent "structural" and "chemical" promoter to promote deNOx performance [15]. Some researchers have reported that the addition of MoO3 could enhance the adsorption and activation of NH3, which was beneficial to the improvement of the NH3-SCR activity [16]. Tang et al. have developed hexagonal WO3 with Mo framework substitution which resulted in hybridizing W and Mo cations with their bridging oxygen ions, thus making the electron transfers in SCR redox cycles relatively easy and leading to improved catalytic activity [17].
Therefore, in this study, we designed these Mo doped CuO samples which can combine the advantages of both species. It is found that this Mo doped CuO catalyst is more active than pure CuO catalyst even in the presence of SO2. According to the various characterization results, the cause of the promoting effect of Mo has been proposed, including structure, chemical and adsorption properties, and reaction mechanism. The synthesis procedure of Mo doped CuO samples, experimental methods, and characterization techniques are provided in Supporting information in detail.
Firstly, the structure of the Mo doped CuO samples was verified by XRD, Raman, and FE-SEM technologies. As shown in Fig. 1a, the XRD patterns for all these samples can be indexed to monoclinic CuO [18]. However, the (−111) and (111) diffraction peaks broaden gradually after Mo doping. With the increasing of Mo dopant amount, the particle size of samples decreases (Table S1 in Supporting information). Significantly, the positions of main diffraction peaks of Mo doped CuO over the 2θ of 35°−40° shift toward higher angles with increasing of the Mo amount, which may arise from the Mo doping into the CuO lattice to form solid solutions. Since the radius of Cu2+ (0.073 nm) ions is larger than that of Mo6+ (0.059 nm), this difference in the ionic radii of the host and the dopant atom can lead to changes in the lattice parameters of the system (Table S1). Therefore, the Cu2+ ions in the lattice of CuO were substituted by Mo6+ ions, which result in monoclinic distortion. When Mo6+ ions substitute Cu2+ ions, Mo6+ will inevitably form in this system. The oxygen vacancies could be formed, resulting from the requirement of charge compensation [19]. As shown in Fig. S1 (Supporting information), the Raman peaks of these Mo doped CuO samples are similar to that of the pure CuO sample. And no active modes related to secondary phases or impurities are observed, indicating the Mo species may be doped into the CuO lattice. A higher magnification FE-SEM images for the 3% Mo doped CuO sample together with Mo, O, and Cu elemental mapping are also presented in Fig. 1b. It is clear that Mo species were uniformly distributed on CuO. The above-mentioned results infer that Mo6+ ions were uniformly doped into the CuO and substituted the position of Cu2+ ions into the lattice of CuO, leading to the formed Mo-O-Cu isostructural substitution.
The influence of Mo species doped CuO for the NH3-SCR activity was investigated. As the results are shown in Fig. 1c, the undoped CuO and pristine MoO3 show very low NH3-SCR efficiency in the whole temperature range. The highest NOx conversion of the CuO sample is only about 58%, which is achieved at 225 ℃. The pure MoO3 catalyst performs a continuous increase of NOx conversion and exhibits a higher NOx conversion than CuO at above 275 ℃. Interestingly, doping of Mo to CuO catalysts resulted in a significant enhancement of deNOx efficiency in the medium-low temperature range with an obviously broadened operation temperature window. Compared with undoped CuO, the NOx conversion of Mo doped CuO catalysts presents a trend of increasing. Further increasing the amount of Mo species to 3 at%, the NH3-SCR activity continues to be improved. This catalyst shows above 80% NOx conversion and 80% N2 selectivity at temperatures from 175 ℃ to 275 ℃ (Fig. S2 in Supporting information). Besides, this Mo doped CuO catalyst has excellent resistance to high space velocity and improved tolerance of SO2 (Figs. S3 and S4 in Supporting information). Compare to other reported Cu-based oxides catalysts [20, 21], this Mo doped CuO oxides catalyst with low cost is one of the potential candidates for deNOx at the low temperature.
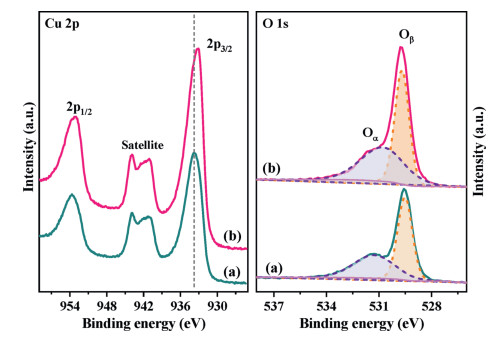
XPS was carried out to study the chemical valence states of the different elements and surface elemental composition in Mo undoped and doped CuO catalysts. Fig. 2 depicts the Cu 2p and O 1s spectra. The peaks at 933.77 eV and 953.65 eV with a spin-energy separation of 19.88 eV can be attributed to Cu 2p3/2 and Cu 2p1/2, which are the characteristics of Cu2+ ions [22]. Furthermore, the appearance of satellite peaks with binding energies of 938.71–947.12 eV further confirms the existence of CuO. The BEs of Cu 2p in this doped sample decrease by 0.61 eV relative to that of Cu 2p in CuO. Depending on the valency, each Mo atom can contribute free electrons to the CuO lattice [23], which could affect the electrical properties of the CuO, inducing the electron density of Cu2+ increment significantly. Therefore, XPS analysis results suggest that the bonding between the Mo and Cu cations indeed occurs, thereby forming the strong electron interaction between Mo and Cu cations. Besides, the optical band gap is obtained on the basis of UV–vis spectra (Fig. S5 in Supporting information). The bandgap value of undoped CuO is 2.31 eV which is similar to other reported CuO [24], while that of Mo doped CuO has been found to be 2.46 eV. Augmenting of the bandgap may be attributed to the charge-transfer transition between Mo6+ d electrons and CuO conduction band [25], which further demonstrates the creation of electron interaction between Cu and Mo in this Mo-O-Cu system. The O 1s peaks were all fitted with two contributions. The O 1s spectra of undoped CuO catalysts present a distinct sharp peak centered around 529.53 eV, which is attributed to the lattice oxygen atoms (Oβ) coordinated with Cu atoms (Cu−O−Cu) [26]. The other shoulder in the higher energy region is related to chemisorbed oxygen species (Oα) [27]. In the case of the Mo doped CuO sample, a slight shift in the BE value of the sharp peak (529.73 eV) is detected, which may be assigned to the appearance of coordination of oxygen in Mo-O-Cu structure. The concentration of Oα is calculated and shown in Table S1. The ratio of Oα species increases with the addition of Mo cations. Based on the XRD and Raman results, it demonstrates that the introduction of Mo dopant is beneficial to the formation of oxygen vacancies, which leads to the amount increase of chemisorbed oxygen species, thereby improving the activity of NO oxidation (Fig. S6a and Fig. S7 in Supporting information). The formed NO2 could enhance the NH3-SCR activity via the "fast-SCR" reaction pathway [28].
The adsorption of NH3 closely related to the surface acidity is crucial for NH3-SCR reaction [29]. The NH3-TPD technology was used to study the surface acidity of the Mo undoped and doped CuO catalysts and their profiles are shown in Fig. 3a. Both undoped and doped CuO samples exhibit a wide desorption peak lasting from 75 ℃ to 400 ℃, indicating the surface acid sites with different thermal stability. The curve of these two catalysts is composed of two portions. The first shoulder peak located below 200 ℃ can be attributed to the physically adsorbed NH3 and weakly bound NH3 species adsorbed on the Lewis or/and Brønsted acid sites, while the second peak at 200–350 ℃ is assigned to the NH3 strongly desorbed on the Lewis or/and Brønsted acid sites [30, 31]. Obviously, the intensity of the NH3 desorption peak becomes higher after the doping of Mo into the CuO catalyst. The adsorption state of NH3 over the 3% Mo doped CuO catalysts surface was characterized by in situ DRIFTs, and the results are shown in Fig. 3b. The bands at 1198 cm−1 are attributed to coordinated NH3 bound to Lewis acid sites, while the broad weak bands that appeared in the region of 1256–1609 cm−1 may be related to the overlap of amide (NH2) species and NH4+ bound to the Brønsted acid sites, respectively [32]. The NH2 species may be derived from the deprotonation of NH3 via partial oxidation which also generates OH species appearing at 3643 and 3614 cm−1 [33]. Based on the above results, the adsorption peak intensity of the Mo doping CuO sample is significantly higher than that of the undoped CuO sample. Combing with the XRD results, the doping of Mo into CuO lattice could result in the formation of Cu-O-Mo structure. It may provide an increased amount of Lewis and Brønsted acid sites, thereby enhancing the NH3 adsorption capacity. On the one hand, the strong electron interaction between Mo and Cu could increase the amount of Lewis acid sites. On the other hand, the formed Mo=O sites could act as Brønsted acid sites on the catalyst surface, since no bands ascribed to NH4+ were observed (Fig. S8 in Supporting information).
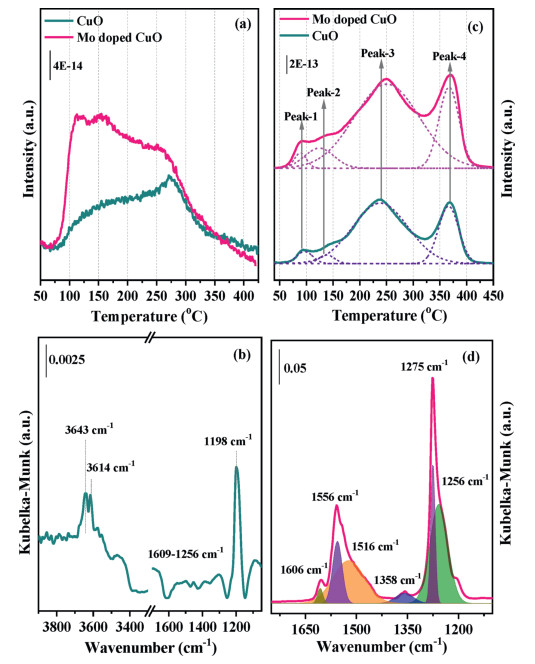
The NOx desorption peaks of CuO and Mo doped CuO catalysts are all fitted with four contributions and are shown in Fig. 3c. The four NOx desorption peaks centered at 94 (peak-1), 138 (peak-2), 238 (peak-3), and 366 ℃ (peak-4) for the undoped CuO sample. The first two weak peaks are ascribed to the desorption of physisorbed NO and NO2 species, respectively [34]. The latter broad peak may be assigned to the decomposition of nitrite or/and monodentate nitrate species, while the last sharp peak could be attributed to the decomposition of thermostable bidentate or bridging nitrate species [35, 36]. After the addition of dopant Mo species, it can be found that the total amount of chemical desorbed NOx increase obviously, especially for NO2 and NO2− or/and monodentate NO3− species (Fig. S9 in Supporting information). The in situ DRIFTS of NOx desorption from 3% Mo doped CuO catalyst were studied, and the results are shown in Fig. 3d. Several bands at 1556, 1516, 1275, 1256, and 1606, 1358 cm−1 appear at 150 ℃ on the Mo doped CuO catalyst. The former three main bands are assigned to the nitrate species, while the band at 1256 cm−1 is a result of the asymmetric deformation of nitrite species (vas(NO2)) [33, 37]. The latter two bands are attributed to gaseous NO2 and monodentate NO2− species, respectively [38].
According to the XRD, Raman, and XPS results, Mo can be doped into CuO structure to form a CuO-liked solid solution containing a Mo−O−Cu bond, leading to a high electron density of Cu ions. On the one hand, it is beneficial to the feasibility of oxygen activation and the oxidation of NO, which have been verified by the XPS and NO oxidation activity results. On the other hand, the redox property is also promoted (Fig. S10 in Supporting information). Compared to the undoped CuO, the reduction peak shifts to the lower temperature, which will be conducive to the NH3 activation (Fig. S6b). In addition, Mo/CuO catalyst was also synthesized by impregnation method and it shows a poor NH3-SCR activity compared to the designed Mo doped CuO catalyst (Fig. S11 in Supporting information). The above-mentioned results manifest that the Mo-O-Cu species with strong interaction between Mo and Cu are the main adsorption and activation sites for NOx and NH3 (Figs. 3a-d), thereby enhancing the NH3-SCR activity.
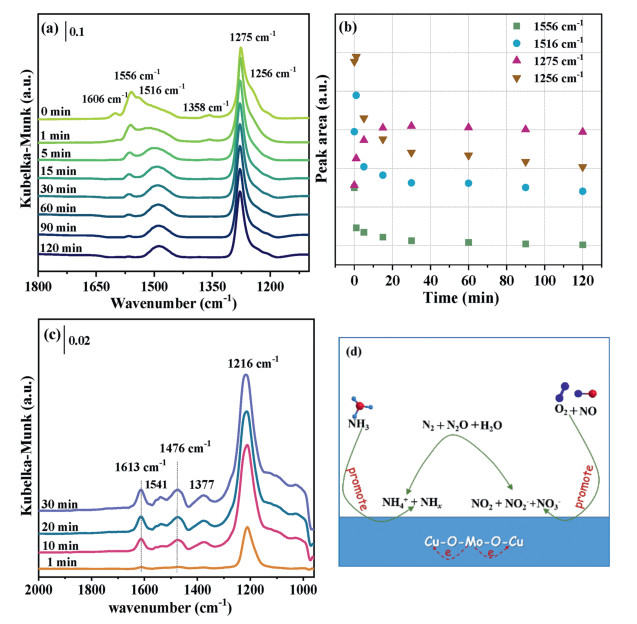
The transient reaction studies were performed by in situ DRIFTS to investigate the reactivity of the NOx and NH3 adsorbed species, thereby exploring the mechanism of NH3-SCR reaction over 3% Mo doped CuO catalyst. The sample was covered by several adsorbed NOx species after pre-adsorption with 500 ppm NO + 3 vol% O2 at 150 ℃ (Fig. 3d). Followed by switching the 500 ppm NH3/He into the cell (after He purged), the bands assigned to NO2 and NO2− rapidly disappeared (Fig. 4a), suggesting the NO2 can react with NHx species via a "fast SCR" reaction pathway. However, the variation trend of the other four peaks over time is slightly different. The area of these peaks was recorded as the function of time and is shown in Fig. 4b. The amount of nitrate species (1556 cm−1) monotonously decrease to a certain value, when ammonia was fed into the sample cell within 2 h, indicating NO3− ad-species are also activated which could react with NH3 ad-species. Interestingly, the peak area of surface NO3− (1516, 1275, and 1256 cm−1) ad-species increases rapidly and then decreases gradually. This may be because intermediate species were generated and then consumed. The FTIR spectrum with pre-adsorbed NH3 exposed to 500 ppm NO + 3 vol% O2 at 150 ℃ is presented in Fig. S12 (Supporting information). After the admission of NOx, the bands assigned to NOx adsorbed species immediately develop and overlap the bands attributed to NH3 ad-species, which suggests that the adsorption capacity of NOx is superior to that of NH3. The spectra of this catalyst at 150 ℃ in the flow of 500 ppm NO + 500 ppm NH3 + 3 vol% O2 were recorded with the purpose of studying the present species under the reaction conditions, which are shown in Fig. 4c. At the initial 1 min, several bands developed at 1612, 1569–1517, 1476, 1377, and 1216 cm−1, which are moderately increased in intensity with increasing the reaction time. The position of these bands shifts, compared with NOx and NH3 adsorption spectra (Fig. S13 in Supporting information), which may result from the formation of intermediate species, e.g., NH4NO3, NH4NO2, NO2(NH3)2 [39]. Meanwhile, the intensity of the bands obtained in the spectra is lower than that in this spectra of NOx adsorption, indicating NOx adsorbed species could react with NH3 ad-species. There may be competitive adsorption between NH3 and NOx. The adsorption sites occupied by NOx results in inhibition of NH3 adsorption and activation. Based on the present investigation, the possible NH3-SCR reaction pathways on the Mo doped CuO catalysts are proposed and shown in Fig. 4d. The gaseous NH3 molecules are first adsorbed on this catalyst to form dominant coordinated NH3 ad-species and a small amount of NH2 and NH4+ ad-species. NO molecules are also adsorbed on the catalyst and then oxidized to NO2, nitrite, and nitrate species. The NO2 can fleetly react with coordinate NH3 ad-species through the E-R mechanism, while the nitrite and nitrate ad-species can react with NH4+ and coordinate NH3 ad-species through the L-H mechanism.
In summary, Mo doped CuO catalyst has been successfully synthesized, which shows an excellent low-temperature NH3-SCR activity. The doping of Mo into CuO lattice leads to the formation of Cu-O-Mo system with strong electron interaction between Cu and Mo, which is beneficial to the oxidation of NO to NO2, thereby enhancing the low-temperature activity via a "fast-SCR" pathway. Moreover, it can also provide more adsorbed sites, improving the adsorption of NH3 and NOx to form coordinate NH3, NH2, NH4+, and NO2−, NO3− ad-species. On the one hand, the NO2 can fleetly react with coordinate NH3 ad-species through the E-R mechanism. On the other hand, the nitrite and nitrate ad-species can react with NH4+, coordinate NH3 and NH2 ad-species through the L-H mechanism. The NOx conversion increases by about 60% at 150 ℃ after the addition of Mo species.
The article has no conflict of interest.
This work was financially supported by the National Natural Science Foundation of China (Nos. 21806017, 21876019), the Fundamental Research Funds for the Central Universities (No. DUT20RC(4)003) and National Key Research and Development Program of China (No. 2019YFC1903903).
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.01.075.
L. Kang, L. Han, J. He, et al., Environ. Sci. Technol. 53 (2019) 938–945. doi: 10.1021/acs.est.8b05637
J.K. Lai, I.E. Wachs, ACS Catal. 8 (2018) 6537–6551. doi: 10.1021/acscatal.8b01357
R. Wu, L. Li, N. Zhang, et al., Catal. Today 376 (2021) 302–310. doi: 10.1016/j.cattod.2020.04.051
L. Zhu, Z. Zhong, H. Yang, C. Wang, J. Environ. Sci. 56 (2017) 169–179. doi: 10.1016/j.jes.2016.08.025
H. Wang, Z. Qu, S. Dong, et al., Environ. Sci. Technol. 50 (2016) 13511–13519. doi: 10.1021/acs.est.6b03589
L. Yan, Y. Liu, H. Hu, et al., ChemCatChem 8 (2016) 2267–2278. doi: 10.1002/cctc.201600332
J. Yu, F. Guo, Y. Wang, et al., Appl. Catal. B: Environ. 95 (2010) 160–168. doi: 10.1016/j.apcatb.2009.12.023
H. Wang, Z. Qu, H. Xie, et al., J. Catal. 338 (2016) 56–67. doi: 10.1016/j.jcat.2016.02.009
N. Zhang, L. Li, B. Zhang, et al., J. Environ. Chem. Eng. 7 (2019) 103044. doi: 10.1016/j.jece.2019.103044
L. Chen, Z. Si, X. Wu, D. Weng, ACS Appl. Mater. Interfaces 6 (2014) 8134–8145. doi: 10.1021/am5004969
Q. Liu, Z. Liu, J. Su, Catal. Today 158 (2010) 370–376. doi: 10.1016/j.cattod.2010.04.036
D. Pietrogiacomi, A. Magliano, D. Sannino, et al., Appl. Catal. B: Environ. 60 (2005) 83–92. doi: 10.1016/j.apcatb.2005.02.025
B. Xu, Y. Liu, Y. Shen, S. Zhu, RSC Adv. 8 (2018) 2586–2592. doi: 10.1039/C7RA12153A
Z. Si, D. Weng, X. Wu, et al., J. Catal. 271 (2010) 43–51. doi: 10.1016/j.jcat.2010.01.025
L. Li, W. Tan, X. Wei, et al., Catal. Commun. 114 (2018) 10–14. doi: 10.1145/3231541.3231545
Z. Liu, J. Zhu, S. Zhang, et al., Catal. Commun. 46 (2014) 90–93. doi: 10.1016/j.catcom.2013.11.032
Y. Chen, Z. Dong, Z. Huang, et al., Catal. Sci. Technol. 7 (2017) 2467–2473. doi: 10.1039/C7CY00416H
J. He, Q. Zhai, Q. Zhang, et al., J. Catal. 299 (2013) 53–66. doi: 10.1016/j.jcat.2012.11.032
P. Lu, P. Wu, J. Wang, X. Ma, Chem. Phys. Lett. 730 (2019) 297–301. doi: 10.1016/j.cplett.2019.06.029
S. Suárez, J.A. Martín, M. Yates, et al., J. Catal. 229 (2005) 227–236. doi: 10.1016/j.jcat.2004.10.019
J. Jemal, H. Tounsi, K. Chaari, et al., Appl. Catal. B: Environ. 113-114 (2012) 255–260. doi: 10.1016/j.apcatb.2011.11.045
X. Zhang, X. Zhang, L. Song, et al., Int. J. Hydrogen Energy 43 (2018) 18279–18288. doi: 10.1016/j.ijhydene.2018.08.060
W. Lv, L. Li, Q. Meng, X. Zhang, J. Mater. Sci. 55 (2020) 2492–2502. doi: 10.1007/s10853-019-04129-9
T. Jan, S. Azmat, Q. Mansoor, et al., Mater. Res. Express 6 (2019) 1050a1053. doi: 10.1088/2053-1591/ab3f8c
R. Swapna, M.C. Santhosh Kumar, J. Phys. Chem. Solids 74 (2013) 418–425. doi: 10.1016/j.jpcs.2012.11.003
X. Zhang, H. Wang, L. Meng, et al., ACS Appl. Energy Mater. 3 (2020) 3465–3476. doi: 10.1021/acsaem.9b02537
Y. Wang, Y. Wang, L. Yu, et al., Chem. Eng. J. 368 (2019) 115–128. doi: 10.1016/j.cej.2019.02.174
J. Han, D. Zhang, P. Maitarad, et al., Catal. Sci. Technol. 5 (2015) 438–446. doi: 10.1039/C4CY00789A
R.T. Guo, X. Sun, J. Liu, et al., Appl. Catal. A: Gen. 558 (2018) 1–8. doi: 10.1016/j.apcata.2018.03.028
L. Ma, C.Y. Seo, M. Nahata, et al., Appl. Catal. B: Environ. 232 (2018) 246–259. doi: 10.1016/j.apcatb.2018.03.065
C. Yu, B. Huang, L. Dong, et al., Chem. Eng. J. 316 (2017) 1059–1068. doi: 10.1016/j.cej.2017.02.024
Z. Ma, X. Wu, H. Härelind, et al., J. Mol. Catal. A: Chem. 423 (2016) 172–180. doi: 10.1016/j.molcata.2016.06.023
G. Qi, R.T. Yang, R. Chang, Appl. Catal. B: Environ. 51 (2004) 93–106. doi: 10.1016/j.apcatb.2004.01.023
S. Zhan, H. Zhang, Y. Zhang, et al., Appl. Catal. B: Environ. 203 (2017) 199–209. doi: 10.1016/j.apcatb.2016.10.010
H. Jiang, Q. Wang, H. Wang, et al., ACS Appl. Mater. Interfaces 8 (2016) 26817–26826. doi: 10.1021/acsami.6b08851
L. Ma, Y. Cheng, G. Cavataio, et al., Appl. Catal. B: Environ. 156-157 (2014) 428–437. doi: 10.1016/j.apcatb.2014.03.048
Z. Liu, S. Zhang, J. Li, et al., Appl. Catal. B: Environ. 158-159 (2014) 11–19. doi: 10.3917/maorg.021.0011
L. Liu, Y. Chen, L. Dong, et al., Appl. Catal. B: Environ. 90 (2009) 105–114. doi: 10.1016/j.apcatb.2009.02.021
R.Q. Long, R.T. Yang, J. Catal. 190 (2000) 22-31. doi: 10.1006/jcat.1999.2737

Figure 1 XRD patterns (a), the elemental mapping images (b) and NOx conversion curve during NH3-SCR reaction (c) of the synthesized samples.

Figure 3 NH3-TPD (a) and NOx-TPD profiles (c) of Mo doped CuO samples. The DRIFT spectra of NH3 adsorption (b) and NOx adsorption (d) of 3 at% Mo doped CuO sample at 150 ℃.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

