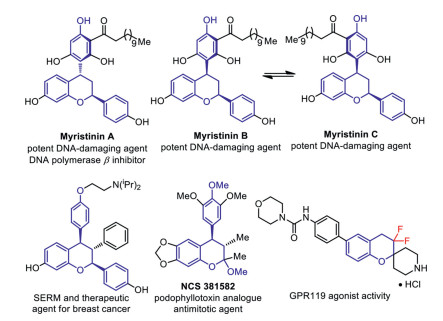
Figure 1.
Representative multisubstituted chroman-containing bioactive molecules.
HFIP-catalyzed highly diastereoselective formal [4 + 2] cyclization to synthesize difluorinated multisubstituted chromans using difluoroenoxysilanes as C2 synthons
Jinshan Li , Wenxue Xi , Saimei Liu , Yaqi Yang , Jianguo Yang , Hanfeng Ding , Zhiming Wang

Chromans, especially multisubstituted chromans have received great attention because of their widely distributed in numerous natural products, pharmaceuticals and bioactive molecules [1]. Representative examples are myristinin A and B/C (potent DNA damaging agents and DNA polymerase β inhibitors) [2], SERM and therapeutic agent for breast cancer [3], NCS 381, 582 (podophyllotoxin analogue, antimitotic agent) (Fig. 1) [4]. Owing to their great value, much effort has been devoted to developing convenient methodologies for the construction of highly functionalized chromans [5, 6]. On the other hand, it is well-known that the selective incorporation of fluorine atoms or fluoroalkyl groups into organic molecules can greatly change the physicochemical and biological properties of the original compounds, such as GPR119 shows agonist activity (Fig. 1) [7, 8]. As a consequence, it is intriguing to develop new protocols for the synthesis of structurally diverse fluorinated chromans. However, the developed synthetic strategies for assembling difluorinated chromans are exceedingly rare and still have a large gap compared with the various well-established approaches for introducing gem-difluoromethylene units (-CF2-, a bioisostere for oxygen or carbonyl group) into a ring-type structure. To date, the conventional methods for the synthesis of difluorinated chromans usually requires multi-step transformation processes via the difluorination of chromone or its derivatives (Scheme 1a) [9]. In view of the above, developing new efficient strategies for facile synthesis of multiply functionalized difluorinated chromans is challenging but highly desirable.
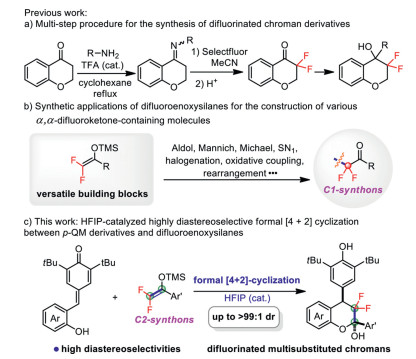
Difluoroenoxysilanes, interesting fluorinated silyl enol ethers, could be easily obtained by the reaction of Mg-mediated C-F bond cleavage of trifluoromethyl ketones, and have been recognized as versatile building blocks for the construction of diverse α, α-difluoroketone-containing molecules [10, 11]. Generally, the reactions of difluoroenoxysilanes mainly involve nucleophilic addition to unsaturated precursors such as aldehydes, ketones, imines and olefins [12-16], nucleophilic substitution of alkyl halides, alcohols or substrates with other leaving groups [17-19], halogenations [20], oxidative coupling [21] and rearrangement reactions [22] lead to numerous α, α-difluoroketone derivatives (Scheme 1b). In spite of elegant achievements, difluoroenoxysilanes usually act as C1 synthons to participate the rapid construction of functionalized α, α-difluoroketones in these reported reaction patterns. Therefore, it is highly desirable to develop new transformation models of difluoroenoxysilanes from the view of expanding the structural diversity of the designed molecules. In order to realize the highly efficient construction of multiply functionalized difluorinated chromans, we envisaged that the generated difluoroketone carbonyl group could be further utilized as an electrophilic site to facilitate the subsequent transformations to forge difluorinated cyclic compounds despite its being rarely explored possibly due to the low reactivity in previous studies [10, 11]. Correspondingly, difluoroenoxysilanes serve as C2 synthons in constructing CF2-engineered chromans for this new reaction pattern. Concerning the excellent performance of ortho-hydroxyphenyl para-quinone methides (p-QMs) in cyclization reactions for synthesizing chroman derivatives [23-29] and our continued interest in developing new catalytic routes to fluorine-containing molecules [16, 18, 19, 22], we herein reported a HFIP-catalyzed highly diastereoselective synthesis of difluorinated multisubstituted chromans via the formal [4 + 2] cyclization of ortho-hydroxyphenyl p-QMs with difluoroenoxysilanes (Scheme 1c). By taking advantage of this scheduled procedure, a series of highly functionalized difluorinated chromans were efficiently prepared in high diastereoselectivities.
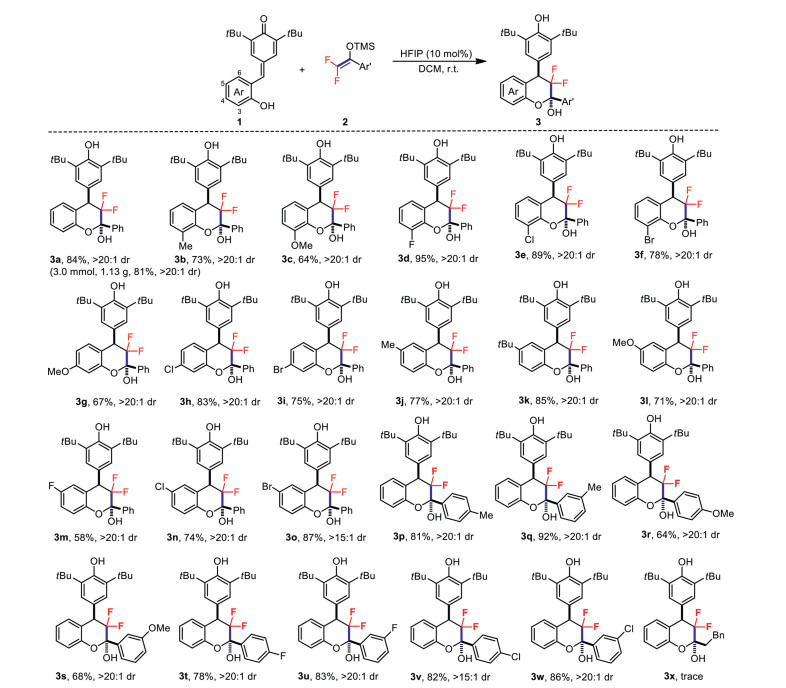
First, ortho-hydroxyphenyl p-QM 1a and difluoroenoxysilane 2a were chosen as the model substrates to examine the feasibility of the envisaged formal [4 + 2] cyclization reaction (Table 1). It was found that the common metal salt Lewis acid catalysts such as Zn(OTf)2, Cu(OTf)2, Sc(OTf)3 and Fe(OTf)3 could promote this formal [4 + 2] cyclization reaction providing the desired highly functionalized difluorinated chroman 3a with excellent diastereoselectivity (entries 1–4, > 20:1 dr). However much lower catalytic efficiency of metal triflate catalysts were observed in these reactions compared with previous reported highly efficient Fe(Ⅲ)-catalyzed 1, 6-conjugate addition of difluoroenoxysilanes to non-ortho-hydroxyphenyl-substituted p-QMs [30], which clearly indicated that the free phenolic OH had an important effect on the 1, 6-addition process (see the control experiments in for more details). Based on these results, we turned our attention to different types of Lewis acids or Brønsted acids, such as B(C6F5)3, Tf2NH, TfOH, PTSA and PhCO2H were evaluated but did not provide a better result (entries 5–9). Inspired by the superior catalytic performance of HFIP for OH-containing substrates in the construction of fluorinated molecules in our previous studies [18, 19], HFIP was then considered as a promising alternative to access the difluorinated chroman products more effectively. As expected, the desired cyclization product 3a was obtained smoothly in high yield with excellent diastereoselectivity (entry 10, 84% yield, > 20:1 dr). Further investigations showed that the multiple fluorine atoms and the solvent had a great influence on the reaction (entries 11–16). Thus the optimal reaction conditions for the formal [4 + 2] cyclization reaction were determined by using 10 mol% HFIP as the catalyst and DCM as the solvent at room temperature.
With the optimized reaction conditions in hand, we next investigated the substrate scope of ortho-hydroxyphenyl p-QMs and difluoroenoxysilanes to probe the generality of this formal [4 + 2] cyclization reaction (Scheme 2). First, ortho-hydroxyphenyl p-QMs with both electron-donating and electron-withdrawing substituents at the 3-position of the phenol ring 1b‒1f were employed. It was obviously found that ortho-hydroxyphenyl p-QMs bearing electron-withdrawing groups afforded the desired difluorinated chroman products (3d‒3f) in a much higher yield than the substrates bearing electron-donating groups (3b and 3c), which was probably promoted by the relative fast proton-transfer process with electron-withdrawing substituents at the ortho-position of bottom phenolic OH group (see the proposed reaction mechanism for details). For substrates with substituents at the 4-position 1g‒1i, the introduction of electron-donating groups also gave a lower yield. While for substrates with substituents at the 5-position 1j‒1o, the introduction of a strong electron-withdrawing group dramatically reduced the reaction yield (3m, 58% yield) (maybe due to the weak nucleophilicity of the resulting phenol ion in subsequent cyclization process). Then, the generality of difluoroenoxysilanes was investigated. Aromatic difluoroenoxysilanes with both electron-donating and electron-deficient groups also gave corresponding difluorinated chroman products 3p−3w in 64%‒92% yields. However, almost no desired cyclization product 3x was detected for benzyl-derived difluoroenoxysilane. Finally, the relative configuration of compound 3a was unambiguously determined by X-ray crystallography analysis (for details, see Supporting information).
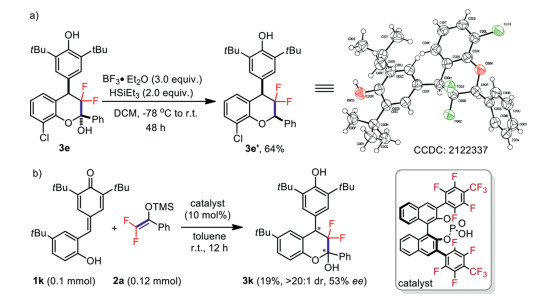
To demonstrate the synthetic utility of this HFIP-catalyzed formal [4 + 2] cyclization reaction, a gram-scale synthesis was conducted. As shown in Scheme 2, the desired difluorinated chroman 3a was obtained in a maintained level of yield and diastereoselectivity. Furthermore, the dehydroxylated structural product 3e′ which is widely exist in bioactive molecules showed in Fig. 1 could also be smoothly obtained with high diastereoselectivity by reductive dehydroxylation of 3e (Scheme 3a). Of particular interest, during the following investigations we found that these difluorinated hemiacetals were perfectly stable under acidic conditions, which shows the great difference between fluorinated and non-fluorinated hemiacetals (the relative configuration of compound 3e′ was determined by X-ray crystallography analysis and studies on the properties of difluorinated multisubstituted chromans, see Supporting information for details). Subsequently, great efforts had been made to developing a catalytic asymmetric variant of this formal [4 + 2] cyclization reaction. It was found that product 3k could be obtained in 19% yield with > 20:1 dr and 53% ee with multiple attempts in the presence of heptafluoro-p-tolyl-substituted chiral monophosphoric acid (Scheme 3b). Though the result is not satisfactory, it also provided a new insight into the development of asymmetric formal [4 + 2] cyclization reaction with difluoroenoxysilanes. Further efforts are underway to develop the catalytic asymmetric version of this reaction with newly designed chiral fluoroalcohol catalysts and the results will be reported in due course.
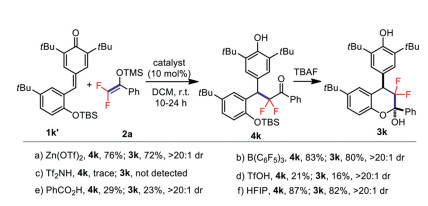
Control experiments were also carried out to gain insight into the reaction mechanism of this cyclization process (Scheme 4). The experiments with TBS-protected substrate 1k' showed that the densely functionalized difluorinated chromans were obtained via a sequential 1, 6-conjugate addition of ortho-hydroxyphenyl p-QMs with difluoroenoxysilanes and subsequent hemiacetalization of the resulting intermediates. Obviously quite different catalytic performance of the used catalysts was observed in this stepwise conjugate addition process, which indicated that the OH group plays a key role in the HFIP-catalyzed formal [4 + 2] cyclization reaction of ortho-hydroxyphenyl p-QMs and the excellent diastereoselectivity of the final products was induced by the special skeleton of the addition intermediates.
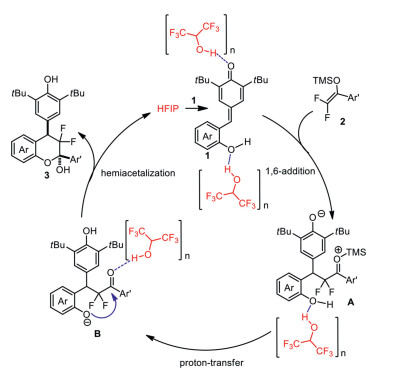
Based on these experiments results and related references [31-37], a proposed mechanism is illustrated in Scheme 5 to explain the reaction process. First, the carbonyl group of ortho-hydroxyphenyl p-QMs 1 is activated by HFIP through hydrogen-bond interaction. On the other hand, the hydrogen-bond interaction between phenolic OH of p-QMs and HFIP may help to improve the reactivity of ortho-hydroxyphenyl p-QMs in 1, 6-addition process. Then, the 1, 6-conjugate addition with difluoroenoxysilanes 2 smoothly affords the desired intermediate A. Finally, after a proton-transfer process and subsequent hemiacetalization, the desired difluorinated chromans 3 are obtained along with the release of HFIP catalyst. It should be noted that the activation potential of HFIP on difluoroenoxysilanes in this formal [4 + 2] cyclization reaction should not be completely ruled out according to our previous study [18].
In summary, an efficient direct formal [4 + 2] cyclization of ortho-hydroxyphenyl p-QMs and difluoroenoxysilanes via HFIP catalysis has been developed. This novel metal-free strategy provides a convenient method to access a variety of multiply functionalized difluorinated chromans with high to excellent diastereoselectivities. By using this method we successfully demonstrated that difluoroenoxysilanes can be commendably applied as C2 synthons, which would bring new opportunities to access structurally diverse cyclic difluorinated molecules efficiently.
The authors report no declarations of interest.
We gratefully acknowledge the financial support from the Natural Science Foundation of Zhejiang Province (No. LQ20B020005), Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005), and Joint Fund of Zhejiang Provincial Natural Science Foundation (No. LTZ21B020001).
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.002.
H.C. Shen, Tetrahedron 65 (2009) 3931-3952. doi: 10.1016/j.tet.2009.02.002
D.J. Maloney, S. Chen, S.M. Hecht, Org. Lett. 8 (2006) 1925-1927. doi: 10.1021/ol060511b
S. Kumar, S. Deshpande, V. Chandra, et al., Bioorg. Med. Chem. 17 (2009) 6832-6840. doi: 10.1016/j.bmc.2009.08.034
J.K. Batra, G.J. Kang, L. Jurd, et al., Biochem. Pharmacol. 37 (1988) 2595-2602. doi: 10.1016/0006-2952(88)90251-1
A. Gaspar, M.J. Matos, J. Garrido, et al., Chem. Rev. 114 (2014) 4960-4992. doi: 10.1021/cr400265z
Z. Sun, T. Zha, Z. Shao, Green Synth Catal. 2 (2021) 241-245. doi: 10.1016/j.gresc.2021.04.002
C. Ni, M. Hu, J. Hu, Chem. Rev. 115 (2015) 765-825. doi: 10.1021/cr5002386
J. Xu, Z. Kuang, Q. Song, Chin. Chem. Lett. 29 (2018) 963-968. doi: 10.1016/j.cclet.2017.10.027
K. Liao, Y. Gong, R. Zhu, et al., Angew. Chem. Int. Ed. 60 (2021) 8488-8493. doi: 10.1002/anie.202016286
M. Decostanzi, J.M. Campagne, E. Leclerc, Org. Biomol. Chem. 13 (2015) 7351-7380. doi: 10.1039/C5OB00855G
X.S. Hu, J.S. Yu, J. Zhou, Chem. Commun. 55 (2019) 13638-13648. doi: 10.1039/C9CC07677H
J.S. Yu, F.M. Liao, W.M. Gao, et al., Angew. Chem. Int. Ed. 54 (2015) 7381-7385. doi: 10.1002/anie.201501747
J.S. Li, Y.J. Liu, G.W. Zhang, et al., Org. Lett. 19 (2017) 6364-6367. doi: 10.1021/acs.orglett.7b03213
X.S. Hu, J.X. He, S.Z. Dong, et al., Nat. Commun. 11 (2020) 5500. doi: 10.1038/s41467-020-19387-4
M.Y. Rong, J.S. Li, F.G. Zhang, et al., Org. Lett. 22 (2020) 9010-9015. doi: 10.1021/acs.orglett.0c03406
J. Li, S. Liu, R. Zhong, et al., Org. Lett. 23 (2021) 5859-5864. doi: 10.1021/acs.orglett.1c01993
X. Gao, R. Cheng, Y.L. Xiao, X.L. Wan, X. Zhang, Chem 5 (2019) 2987-2999. doi: 10.1016/j.chempr.2019.09.012
J. Li, W. Xi, R. Zhong, et al., Chem. Commun. 57 (2021) 1050-1053. doi: 10.1039/D0CC06980A
J. Li, W. Xi, S. Liu, et al., Org. Lett. 23 (2021) 7264-7269. doi: 10.1021/acs.orglett.1c02659
X. Jiang, D. Meyer, D. Baran, et al., J. Org. Chem. 85 (2020) 8311-8319. doi: 10.1021/acs.joc.0c01030
L. Chu, X. Zhang, F.L. Qing, Org. Lett. 11 (2009) 2197-2200. doi: 10.1021/ol900541n
J. Li, Y. Chen, R. Zhong, et al., Org. Lett. 22 (2020) 1164-1168. doi: 10.1021/acs.orglett.0c00018
J.Y. Wang, W.J. Hao, S.J. Tu, et al., Org. Chem. Front. 7 (2020) 1743-1778. doi: 10.1039/D0QO00387E
C.G.S. Lima, F.P. Pauli, D.C.S. Costa, et al., Eur. J. Org. Chem. 2020 (2020) 2650-2692. doi: 10.1002/ejoc.201901796
P. Chauhan, U. Kaya, D. Enders, Adv. Synth. Catal. 359 (2017) 888-912. doi: 10.1002/adsc.201601342
K. Zhao, Y. Zhi, T. Shu, et al., Angew. Chem. Int. Ed. 55 (2016) 12104-12108. doi: 10.1002/anie.201606947
S. Liu, X.C. Lan, K. Chen, et al., Org. Lett. 19 (2017) 3831-3834. doi: 10.1021/acs.orglett.7b01705
C. Duan, L. Ye, W. Xu, et al., Chin. Chem. Lett. 29 (2018) 1273-1276. doi: 10.1016/j.cclet.2017.11.044
L. Zhang, H. Yuan, W. Lin, et al., Org. Lett. 20 (2018) 4970-4974. doi: 10.1021/acs.orglett.8b02088
Y.J. Hao, X.S. Hu, J.S. Yu, et al., Tetrahedron 74 (2018) 7395-7398. doi: 10.1016/j.tet.2018.11.017
X.D. An, J. Xiao, Chem. Rec. 20 (2020) 142-161. doi: 10.1002/tcr.201900020
C.J. Yu, J. Sanjose-Orduna, F.W. Patureau, et al., Chem. Soc. Rev. 49 (2020) 1643-1652. doi: 10.1039/C8CS00883C
V. Pozhydaiev, M. Power, V. Gandon, et al., Chem. Commun. 56 (2020) 11548-11564. doi: 10.1039/D0CC05194B
T. Bhattacharya, A. Ghosh, D. Maiti, Chem. Sci. 12 (2021) 3857-3870. doi: 10.1039/D0SC06937J
A. Berkessel, J.A. Adrio, J. Am. Chem. Soc. 128 (2006) 13412-13420. doi: 10.1021/ja0620181
I. Colomer, ACS Catal. 10 (2020) 6023-6029. doi: 10.1021/acscatal.0c00872
C. Qi, G. Force, V. Gandon, et al., Angew. Chem. Int. Ed. 60 (2021) 946-953. doi: 10.1002/anie.202010846

Scheme 1 State of the art for construction difluorinated multisubstituted chromans and our work design.

Scheme 2 Substrate scope. Reaction conditions: 1 (0.5 mmol), 2 (0.6 mmol), HFIP (10 mol%), DCM (3.0 mL), room temperature, 10–36 h. Isolated yield. Diastereomeric ratios were determined by NMR analysis of the crude reaction mixture.

Scheme 3 Synthetic transformation and preliminary investigations toward the catalytic asymmetric variant.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

