cjli@shu.edu.cn (C. Li). 1 These two authors contributed equally to this work.
Received Date:
09 March 2021 Accepted Date:
29 March 2021 Revised Date:
26 March 2021 Available Online:
01 October 2021
Abstract:
Advanced chemotherapy strategies are in urgent demand for improving anticancer efficacy. Herein, a water-soluble pillar[6]arene (WP6A) was used to load chemotherapeutic agent pemetrexed (PMX) by forming direct host-guest inclusion, which is beneficial for decreasing cytotoxicity of PMX on BEAS-2B cells. NMR and florescence titration served to confirm the complexation between WP6A and ATP with higher affinity [(5.67 ± 0.31) × 105 L/mol], favoring competitive replacement of PMX. Complexation ATP by WP6A effectively prevented ATP from being hydrolyzed in presence of alkaline phosphatase. The formed host-guest complex was further used to block the efflux pump by cutting off energy source from ATP hydrolysis, which was accompanied with releasing PMX to produce synergistic enhancement of anticancer performance towards A549 cells. This supramolecular strategy would also be extended to other clinical chemotherapeutic agents and it was expected to provide salutary profits for cancer patients.
Cancer signals a leading cause of mortality over the world and the patient number is forecast to reach over 21 million by 2030 [1]. Though considerable efforts have been made for treatment of cancer, chemotherapy is still the most important approach in clinic [2-4]. However, traditional chemotherapy faces lots of challenges in practical application, such as serious side effects and ambiguous anticancer efficacy [5-7]. Supramolecular chemotherapy originated from the marriage between host-guest chemistry and chemotherapy, provides an effective and readily generalizable strategy for enhancing anticancer bioactivity of chemotherapy agents against cancer cells and decreasing cytotoxicity to normal cells [8-10]. The key to this strategy lies in one of the cancer biomarkers, which is overexpressed in cancerous microenvironments. Biomarker-triggered release of anticancer agents and simultaneous consumption of biomarker via noncovalent interactions can achieve synergistic improvement of therapeutic effect [11-14]. Adenosine 5'-triphosphate (ATP), one of the most important biomolecules in living organisms, is seen as direct source of energy for various life activities [15, 16]. Cells produce ATP through two related mechanisms, namely that oxidative phosphorylation in mitochondria and glycolysis in the cytosol [17-19]. Compared with normal cells, cancerous cells are characterized by a marked increase in glycolytic metabolism, leading to significant elevation in ATP level [20, 21]. Efflux transporter, like P-glycoprotein (P-gp) can utilize the energy from ATP hydrolysis to decrease intracellular levels of anticancer agents below their effective dose, even leading to multidrug resistance [22-24]. Cutting off the energy source by host-guest encapsulation using artificial receptors was expected to block the efflux pump [25, 26]. Given that, ATP can be identified as a feasible biomarker in this work.
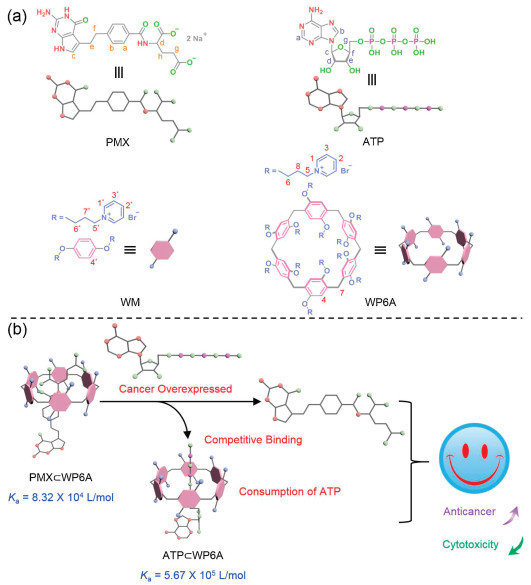
As a proof of concept, pemetrexed (PMX), the first-line chemotherapeutic agent for lung adenocarcinoma, served as model anticancer agent [27-29]. A water-soluble pillar[6]arene (WP6A) containing multiple pyridinium moieties was prepared as macrocyclic receptor, affording strong binding to PMX as well as ATP (Fig. 1) [30-42]. Strong binding to PMX is prerequisite to avoid its serious damage against normal cells. Then considerable binding to ATP is necessary to trigger the PMX release. The formation of a stable host-guest inclusion complex ATP/WP6A was found to efficiently inhibit the hydrolysis of ATP in the presence of alkaline phosphatase. High concentration of ATP in cancerous microenvironments was conducive to replacing of PMX from PMX/WP6A. As a consequence, the efficacy of chemotherapy of PMX was significantly improved against human lung adenocarcinoma epithelia cells and cytotoxicity toward human normal pulmonary epithelial cells has receded.
Figure 1
Figure 1.
(a) Chemical structures of PMX, ATP, macrocycle monomer (WM) and WP6A. (b) Schematic illustration of chemotherapy strategy based on host-guest complexation between PMX and WP6A and release of PMX from WP6A via competitive binding to ATP.
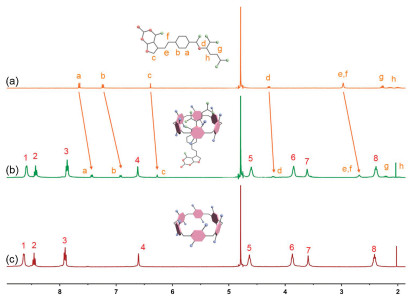
1H NMR spectroscopy was employed first to verify host-guest complexation between host (WP6A) and guestes (PMX and ATP). Figs. 2a-c show the 1H NMR spectra of PMX in deuterated phosphate buffer (pD 7.4) in the absence and presence of WP6A. Upon addition of one equivalent of WP6A, the resonance peaks related to protons Ha, Hb, He and Hf of PMX exhibited remarkable upfield shifts and broadening effects comparing to free PMX (Δδ -0.22 ~ -0.31) suggesting that these protons were engulfed by the cavity of WP6A and were shielded by the electron-rich cyclic structure by forming an inclusion complex. In additon, the host shows similar binding behaviors toward ATP (Fig. S9 in Supporting information). The proton signals for Ha, Hb and Hc of ATP underwent substantial upfield shift and broadening effects, indicating that ATP was partially located in the cavity of WP6A. For comparison, controlled 1H NMR experiments of mixtures of macrocycle monomer (WM, 6 equv.) and two guests (PMX and ATP) were also measured (Figs. S10 and S11 in Supporting information), showing that PMX and ATP could not interact with WM.
Figure 2
Figure 2.1H NMR spectra (400 MHz, 298 K) of (a) PMX (2.0 mmol/L), (b) PMX (2.0 mmol/L) + WP6A (2.0 mmol/L) and (c) WP6A (2.0 mmol/L) in the 20 mmol/L phosphate buffer solution (pD 7.4).
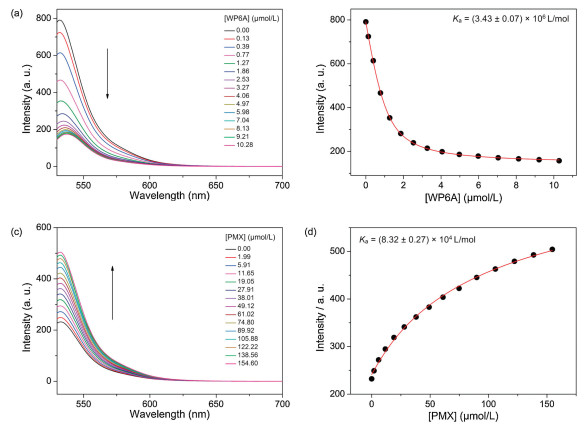
The binding affinities of WP6A with two guests (PMX and ATP) were determined using a fluorescent indicator dispalcement assay [43-45] with Eosin Y (EY) as the optimal reporter dye. The complexation stoichiometry between WP6A and EY was verified to be 1:1 by the continuous variation method (Job's plot method) result using the fluorescence emission values at 538nm (Fig. S12 in Supporting information). EY was strongly complexed by WP6A with a binding constant of Ka = (3.43 ± 0.07) × 106 L/mol, which was ideal for the projected competitive titrations (Figs. 3a and b). The displacement of EY/WP6A by gradual addition of PMX resulted in regeneration of the instrinsic emission of EY (Fig. 3c). The data were well fitted by a 1:1 competitive binding model, giving a Ka value of (8.32 ± 0.27) × 104 L/mol (Fig. 3d). Robust binding to PMX would be prerequisite to avoid unwarranted off-target leaking during delivery process. By comparison with the Ka value of ATP/WP6A [(5.67 ± 0.31) × 105 L/mol, Fig. S13 in Supporting information], such a high binding constant would endow ATP with an advantage to competitively bind to WP6A for thoroughly releasing PMX from the PMX/WP6A complex.
Figure 3
Figure 3.
(a) Direct fluorescence titration of EY (1.0 μmol/L) with WP6A in 10 mmol/L HEPES buffer at pH 7.4, λex = 517 nm. (b) The associated titration curve at λem = 538 nm and fit according a 1:1 binding stoichiometry based on the concentration of WP6A. (c) Competitive fluorescence titration of PMX in the presence of EY (1.0 μmol/L) and WP6A (2.5 μmol/L) in 10 mmol/L HEPES buffer at pH 7.4, λex = 517 nm. (d) The associated competitive titration curve at λem = 538 nm and fit according a 1:1 competitive binding model. Data are from n = 3 independent experiments and are presented as mean ± SD.
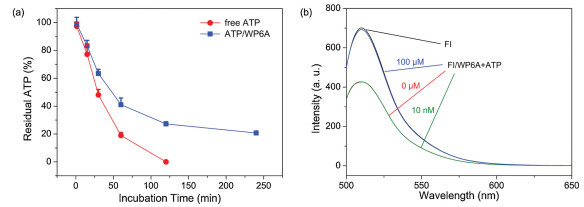
Previous studies have shown that the hydrolysis of ATP could be inhibited by host-guest inclusion using macrocyclic receptors [25, 26]. Drug efflux pump of cancer cells, like P-gp protein, can utilize the energy from ATP hydrolysis to transport anticancer agents out resulting in multidrug resistence [22-24]. The design of this chemotherapy strategy was expected to provide a benefit in therapeutic effect via competitive binding between ATP and WP6A. To test this hypothesis, the hydrolysis rate of ATP and 1:1 mixture of ATP/WP6A in presence of alkaline phosphatase (CIAP) was studied using high-performance liquid chromatography (HPLC). First, appropriate calibration curve was derived (Fig. S14 in Supporting information). As shown in Fig. 4a, unpon exposure to CIAP for 120 min, free ATP underwent rapid hydrolysis and became undetectable. In the sharp contrast, ATP/WP6A retained (20.73 ± 0.82)% of original amount for even 240 min. The above phenomenon occurred because ATP was engulfed in the undisturbed cavity of WP6A preventing ATP from being hydrolyzed.
Figure 4
Figure 4.
(a) ATP (1.0 μmol/L) hydrolysis in the presence of alkaline phosphatase (CIAP, 10 μg/mL). (b) Fluorescence spectra of FI in the absence (black) and presence of 4.0 μmol/L WP6A (red), and upon addition of 100 μmol/L ATP (blue) and 10 nmol/L ATP (green) in 10 mmol/L HEPES buffer at pH 7.4.
The ATP-triggered release was then investigated via an indicator displacement assay. Fluorescein (FI) was screened as reporter dye since Ka for FI/WP6A was comparative to that of PMX/WP6A at the same order of magnitude (Fig. S16 in Supporting information). Adding WP6A to FI solution gave rise to the fluorescence quenching (Fig. 4b). ATP was severely overexpressed in cancer microenvironment ([ATP] < 100 μmol/L) than in normal tissues ([ATP] < 10 μmol/L) [46-48]. Therefore 10 nmol/L and 100 μmol/L ATP were employed to reflect those of normal physiological environment and cancer microenvironment respectively. The intrinsic emission of FI was almost completely regenerated at 100 μmol/L ATP, while negligible change of fluorescence at 10 nmol/L ATP was observed (Fig. 4b). We thus suggested that PMX/WP6A underwent ATP-triggered competitive release of PMX within ATP-overexpressed cancer microenvironment.
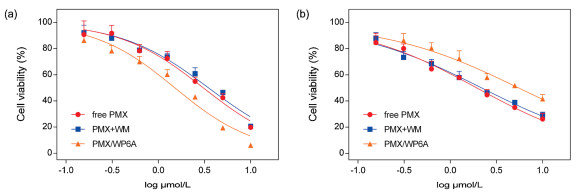
To verify the above chemotherapy strategy could work at the cellular level, the cytotoxicity of WP6A in the human lung adenocarcinoma epithelia cells (A549) and human normal pulmonary epithelial cells (BEAS-2B) were assessed using a Cell Counting Kit-8 (CCK-8) assay. WP6A displayed minimal cytotoxicity against these two cell lines over the testing range (Fig. S17 in Supporting information). These same A549 and BEAS-2B cell lines were then used test the in vitro cell inhibitory effect of PMX/WP6A and two controls (Figs. 5a and b). Concentration-dependent cell death could be seen in all formulations. The half-maximum inhibitory concentration (IC50) value of free PMX for A549 cell line (3.07 μmol/L) matched that recorded in the literature [49-51] and the cytotoxic activity of PMX + WM was nearly equal to that of free PMX over a range of concentrations. While PMX/WP6A exhibited higher anticancer bioactivity than PMX alone. The significant anticancer efficacy improvement was likely attributed to selected release of PMX from complex upon encountering a high concentration ATP inside cancer cells and the energy source of efflux pump was cut off by forming stable inclusion complex ATP/WP6A. In addition, we found interestingly that, via host-guest complexation, the cytotoxicity of PMX/WP6A to BEAS-2B presented significant reduce. The reason was that ATP level in normal cells was frequently lower than that in cancer cells and lower concentration of ATP would be insufficient to cause complete PMX release via competitive replacement.
Figure 5
Figure 5.
Cytotoxicity seen in the (a) A549 and (b) BEAS-2B cell lines incubated with the indicated agents for 48 h. Cell death was measured using CCK-8 assays (mean ± SD, n = 5).
Moreover, from the morphology of living cells we could further confirmed that PMX/WP6A could improve anticancer bioactivity of PMX against A549 cells and decrease its cytotoxicity to BEAS-2B cells (Fig. S18 in Supporting information). In addition, to investigate the apoptosis of A549 cells and BEAS-2B cells, FITC-Annexin V/propidium iodide (PI) method was employed. As shown in Fig. S19 (Supporting information), negligible apoptosis happened in the PBS group in A549 cell line. The percentage of apoptotic cells (including early and late apoptotic cells) in free PMX group was about 24.24%, while that in the presence of PMX/WP6A was significantly higher at 41.59%. For BEAS-2B cell line, the total apoptotic ratios of PMX/WP6A (14.53%) was lower than that of free PMX (19.20%) (Fig. S20 in Supporting information). Taken in concert, the cellular results provided support for the conclusion that inclusion PMX within WP6A could improve the anticancer bioactivity of PMX and decrease its toxic side effects to normal cells.
In summary, we have demonstrated an effective and feasible chemotherapy strategy that involved the direct encapsulation of an anion chemotherapeutic agent, PMX by an appropriate macrocyclic host WP6A. Host-guest complexation served to decrease cytotoxicity of PMX to normal cells. The higher binding affinity of ATP/WP6A is conducive to replacing PMX from its complex. More interestingly, PMX/WP6A exhibited better anticancer activity because the thorough release of PMX by overexpressed ATP and simultaneous comsumption of ATP by WP6A can produce synergistic chemotherapeutic efficacy. This research not only demonstrates that host-guest inclusion is a promising approach to improve treatment effect on cancer but also manifests that water-soluble pillararenes can serve as an ideal platform to endow other clinical agents with benefits.
Declaration of competing interest
The authors report no declarations of interest.
Acknowledgments
We acknowledge the National Natural Science Foundation of China (Nos. 21772118, 21971192, 81573354), and the Natural Science Foundation of Tianjin City (No. 20JCZDJC00200).
Figure 1
(a) Chemical structures of PMX, ATP, macrocycle monomer (WM) and WP6A. (b) Schematic illustration of chemotherapy strategy based on host-guest complexation between PMX and WP6A and release of PMX from WP6A via competitive binding to ATP.
Figure 3
(a) Direct fluorescence titration of EY (1.0 μmol/L) with WP6A in 10 mmol/L HEPES buffer at pH 7.4, λex = 517 nm. (b) The associated titration curve at λem = 538 nm and fit according a 1:1 binding stoichiometry based on the concentration of WP6A. (c) Competitive fluorescence titration of PMX in the presence of EY (1.0 μmol/L) and WP6A (2.5 μmol/L) in 10 mmol/L HEPES buffer at pH 7.4, λex = 517 nm. (d) The associated competitive titration curve at λem = 538 nm and fit according a 1:1 competitive binding model. Data are from n = 3 independent experiments and are presented as mean ± SD.
Figure 4
(a) ATP (1.0 μmol/L) hydrolysis in the presence of alkaline phosphatase (CIAP, 10 μg/mL). (b) Fluorescence spectra of FI in the absence (black) and presence of 4.0 μmol/L WP6A (red), and upon addition of 100 μmol/L ATP (blue) and 10 nmol/L ATP (green) in 10 mmol/L HEPES buffer at pH 7.4.
Figure 5
Cytotoxicity seen in the (a) A549 and (b) BEAS-2B cell lines incubated with the indicated agents for 48 h. Cell death was measured using CCK-8 assays (mean ± SD, n = 5).

 DownLoad:
DownLoad:

 下载:
下载:






