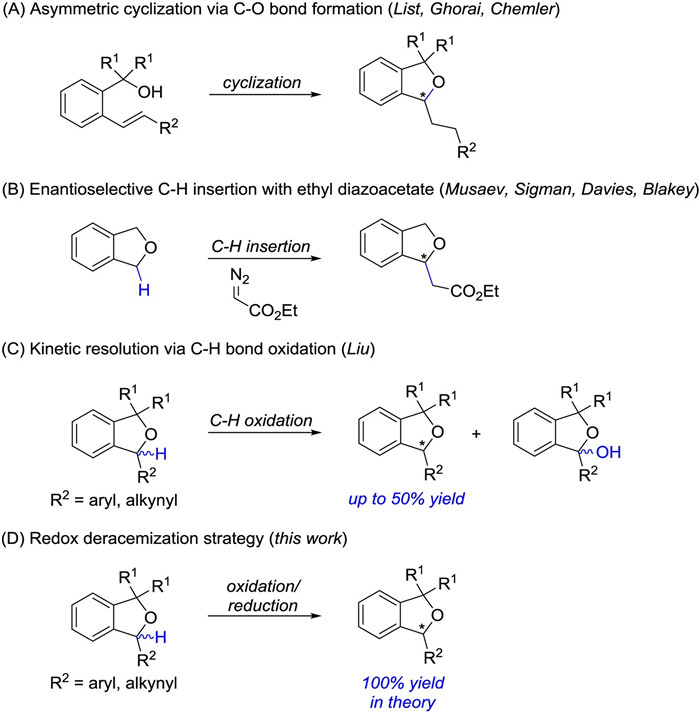
Scheme 1.
Overview of catalytic asymmetric synthesis of α-substituted 1, 3-dihydroisobenzofurans.
Redox deracemization of α-substituted 1, 3-dihydroisobenzofurans
Xiaohan Chen , Ran Zhao , Ziqiang Liu , Shutao Sun , Yingang Ma , Qingyun Liu , Xia Sun , Lei Liu

Chiral α-substituted 1, 3-dihydroisobenzofurans are key scaffolds in a number of bioactive natural products and synthetic pharmaceuticals [1]. Although a variety of racemic routes have been reported, there are surprisingly few catalytic enantioselective approaches to this important class of chiral molecule [2-4]. Existing studies predominantly relied on asymmetric intramolecular cyclization strategy involving C–O bond formation (Scheme 1A) [2]. Despite excellent ee values, pre-installation of reactive functional groups, such as alkenes and ethers, were prerequisite. Subsequently, an elegant iridium(III)-catalyzed enantioselective C–H alkylation of 1, 3-dihydroisobenzofurans with ethyl diazoacetate was disclosed, though 4.0 equiv. of the substrate was required (Scheme 1B) [3]. Recently, our group developed a kinetic resolution of racemic α-aryl and -alkynyl substituted 1, 3-dihydroisobenzofurans through asymmetric C–H oxidation with excellent chiral recognition (Scheme 1C) [4]. However, the theoretical yield of the recovered enantiopure phthalans can not exceed a limit of 50%. Therefore, development of a strategically different approach through C–H bond manipulation in high efficiency would be a highly attractive project to pursue.
Deracemization, wherein a racemic target is directly and completely converted to a single enantiomer of exactly the same molecule in theoretically 100% yield, has emerged as an attractive and straightforward alternative to conventional asymmetric synthesis [5]. Despite conceptual simplicity and potential practical benefits, catalytic non-enzymatic deracemization has remained scarce [6], and existing methods typically focus on alcohol or amine substrates through oxidation and reduction chemistry, whose oxidized intermediates are stable and isolable ketones and imines, respectively [7-9]. Redox deracemization of ethers involving unstable oxidized oxocarbenium ion intermediates remained rarely disclosed, and existing method always focuses on six-membered cyclic benzylic ethers [10]. Herein, we report a one-pot redox deracemization of α-substituted 1, 3-dihydroisobenzofurans (Scheme 1D).
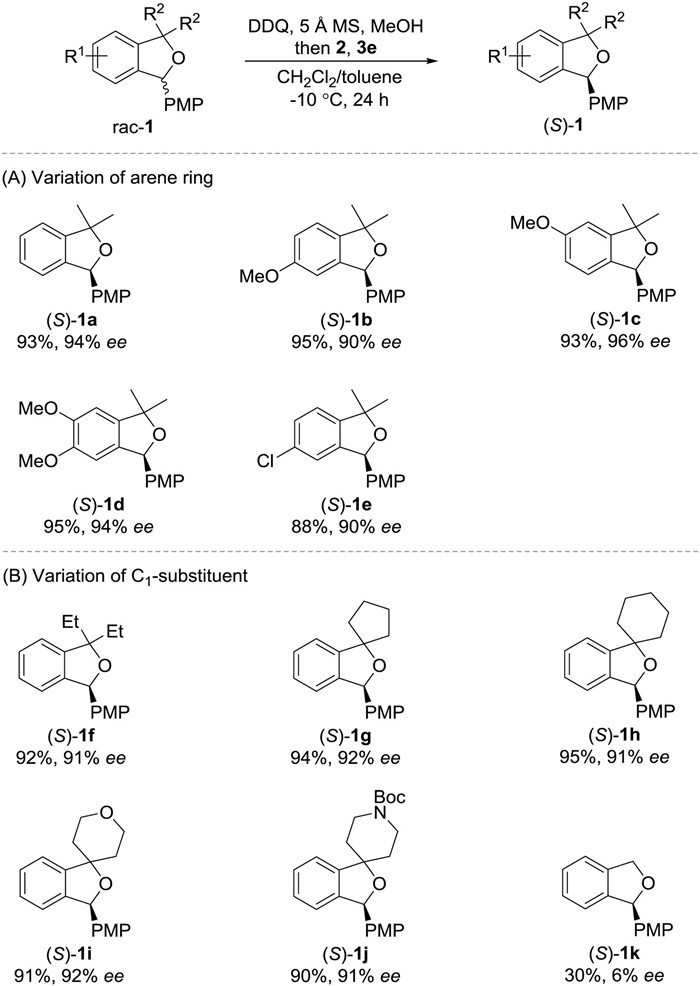
Initially, redox deracemication of 1, 3-dihydroisobenzofuran rac-1a was chosen for optimization employing Hantzsch ester 2 and chiral phosphoric acid (CPA) 3 as the asymmetric reduction system (Table 1) [11]. The reaction was performed in a two-step, one-pot manner to avoid direct quench of oxidant and reductant. When 1 equiv. of DDQ was used as the oxidant, an incomplete consumption of rac-1a was observed (entry 1, Table 1) [12]. Protic additives proved to be crucial to complete oxidation and enantioselectivity, and MeOH was identified to be optimal (entries 2-6). Reaction without 5 Å molecular sieve afforded an inferior result (entry 7). A series of CPA catalysts were then evaluated, revealing that C2-symmetric imidodiphosphoric acid 3e was the best choice, which might be ascribed to the sterically highly demanding and rigid chiral microenvironment around the active site of 3e (entries 3 and 8-12). Solvent optimization suggested that deracemization proceeded most efficiently in a mixed solvents of CH2Cl2 and toluene (entry 13). Adding Hantzsch ester prior to the oxidation was ineffective (entry 14).
With the optimized reaction conditions in hand, the scope of redox deracemization of 1, 3-dihydroisobenzofurans was explored (Scheme 2). A range of electron-donating and -withdrawing substituents on the benzylic ether arene skeleton were well tolerated, as demonstrated by generation of enantiopure 1a-1e in good yields (88%–95%) with excellent enantiocontrol (90%–96% ee) (Scheme 2A). Modification of the geminal C1-disubstitution did not impair the reactivity and selectivity for 1f-1j (Scheme 2B). Notably, spirocyclic 1g-1j bearing diverse ring size and functionality pattern were compatible with the deracemization process. The deracemization of simple 1, 3-dihydroisobenzofuran 1k without geminal C1 disubstitution was next explored. While a complete oxidation could be achieved, a competitive oxidation at C1 position was also observed. The geminal C1 disubstitution also proved to be crucial to the enantioselectivity, and 1k was isolated in 30% yield with 6% ee.
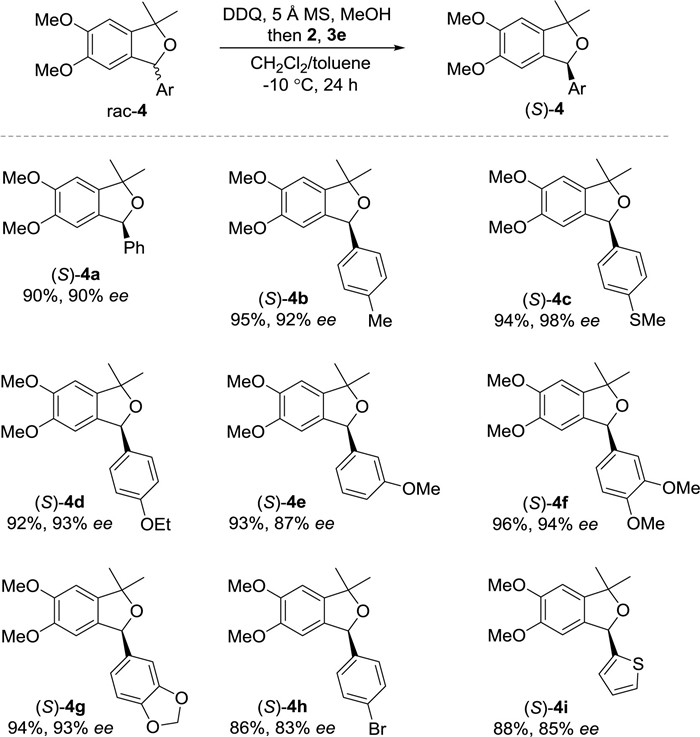
The scope of α-substituent patterns was next investigated (Scheme 3). A variety of electronically varied aryl moieties at α position participated in the redox deracemization smoothly, providing respective 4a-4h in good yields with good to excellent ee (83%-98%). Heteroaryl rac-4i was also a suitable component.
Besides diverse α-aryl substituents, α-alkynyl substituted 1, 3-dihydroisobenzofurans were also competent substrates, as demonstrated by effective deracemization of rac-5 giving (S)-5 in 90% yield with 80% ee (Scheme 4). Under the standard oxidation conditions, α-alkyl substituted 1, 3-dihydroisobenzofurans cannot be completely oxidized (see Supporting information for details).

The additive MeOH proved to be beneficial for reaction efficiency and enantioselectivity. Accordingly, the deracemization process was performed stepwise to understand the role of MeOH. Oxidation of rac-1a with DDQ and MeOH proceeded smoothly, affording ketal 6 in high efficiency (Scheme 5A). Under the standard deracemization conditions in the absence of oxidation elements, ketal 6 was reduced to (S)-1a with comparable ee to that observed in deracemization reaction (Scheme 5B). The observations suggested that a ketal intermediate might be involved in the deracemization process. Based on the results in entries 2-6 in Table 1, 1, 3-dihydroisobenzofuran-based ketal bearing a methoxy moiety might be the best intermediate for asymmetric transfer hydrogenation with respective to the enantiocontrol, though the origin of the observation remains unclear.

Based on the above studies, a plausible mechanism for redox deracemization of 1, 3-dihydroisobenzofurans was suggested (Scheme 5C). Ether rac-1a was oxidized by DDQ to generate oxocarbenium intermediate 7, which was captured by MeOH giving ketal 6. CPA 3e catalyzed the collapse of 6, furnishing a contact ion pair 8 for subsequent asymmetric reduction with Hantzsch ester 2 to complete the redox deracemization process.
In summary, a one-pot redox deracemization of α-substituted 1, 3-dihydroisobenzofurans has been disclosed. MeOH was found to be crucial to the reaction efficiency and enantioselectivity by forming a ketal intermediate. A range of α-aryl substituted five-membered cyclic benzylic ethers were effectively deracemized. α-Alkynyl substituted ethers were also compatible with the deracemization technology.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financial supported by the National Natural Science Foundation of China (Nos. 21971148) and Shenzhen Special Funds (No. JCYJ20190807093805572).
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.021.
(a) V.J. Bauer, B.J. Duffy, D. Hoffman, et al., J. Med. Chem. 19 (1976) 1315-1324;
(b) E.G. Maestrup, C. Wiese, D. Schepmann, et al., Bioorg. Med. Chem. 19 (2011) 393-405.
(a) I. Čorić, J.H. Kim, T. Vlaar, et al., Angew. Chem. Int. Ed. 52 (2013) 3490-3493;
(b) B. Ravindra, B.G. Das, P. Ghorai, Org. Lett. 16 (2014) 5580-5583;
(c) D. Chen, S.R. Chemler, Org. Lett. 20 (2018) 6453-6456;
(d) D. Chen, I.A. Berhane, S.R. Chemler, Org. Lett. 22 (2020) 7409-7414.
N.M. Weldy, A.G. Schafer, C.P. Owens, et al., Chem. Sci. 7 (2016) 3142-3146. doi: 10.1039/C6SC00190D
(a) S. Sun, Y. Ma, Z. Liu, L. Liu, Angew. Chem. Int. Ed. 60 (2021) 176-180;
(b) S. Sun, Y. Yang, R. Zhao, D. Zhang, L. Liu, J. Am. Chem. Soc. 142 (2020) 19346-19353.
(a) K. Faber, Chem. Eur. J. 7 (2001) 5004-5010;
(b) C.C. Gruber, I. Lavandera, K. Faber, W. Kroutil, Adv. Synth. Catal. 348 (2006) 1789-1805;
(c) N.J. Turner, Curr. Opin. Chem. Biol. 14 (2010) 115-121;
(d) M. Rachwalski, N. Vermue, F.P.J.T. Rutjes, Chem. Soc. Rev. 42 (2013) 9268-9282;
(e) R.C. Simon, N. Richter, E. Busto, W. Kroutil, ACS Catal. 4 (2014) 129-143.
(a) A. Hölzl-Hobmeier, A. Bauer, A.V. Silva, et al., Nature 564 (2018) 240-243;
(b) A. Tröster, A. Bauer, C. Jandl, T. Bach, Angew. Chem. Int. Ed. 58 (2019) 3538-3541;
(c) N.Y. Shin, J.M. Ryss, X. Zhang, et al., Science 366 (2019) 364-369.
(a) C.V. Voss, C.C. Gruber, W. Kroutil, Angew. Chem. Int. Ed. 47 (2008) 741-745;
(b) C.V. Voss, C.C. Gruber, K. Faber, et al., J. Am. Chem. Soc. 130 (2008) 13969-13972;
(c) C.J. Dunsmore, R. Carr, T. Fleming, et al., J. Am. Chem. Soc. 128 (2006) 2224-2225;
(d) D. Ghislieri, A.P. Green, M. Pontini, et al., J. Am. Chem. Soc. 135 (2013) 10863-10869;
(e) K. Yasukawa, S. Nakano, Y. Asano, Angew. Chem. Int. Ed. 53 (2014) 4428-4431.
(a) Y. Shimada, Y. Miyake, H. Matsuzawa, et al., Chem. Asian J. 2 (2007) 393-396;
(b) G.R.A. Adair, J.M.J. Williams, Chem. Commun (2007) 2608-2609;
(c) P. Qu, M. Kuepfert, S. Jockusch, et al., ACS Catal. 9 (2019) 2701-2706;
(d) A.D. Lackner, A.V. Samant, F.D. Toste, J. Am. Chem. Soc. 135 (2013) 14090-14093;
(e) Y. Ji, L. Shi, M.W. Chen, et al., J. Am. Chem. Soc. 137 (2015) 10496-10499;
(f) L. Zhang, R. Zhu, A. Feng, et al., Chem. Sci. 11 (2020) 4444-4449.
(a) Y. Mao, Z. Wang, G. Wang, et al., ACS Catal. 10 (2020) 7785-7791;
(b) Y. Ma, X. Liu, Y. Mao, et al., Org. Chem. Front. 7 (2020) 2526-2530.
M. Wan, S. Sun, Y. Li, et al., Angew. Chem. Int. Ed. 56 (2017) 5116-5120. doi: 10.1002/anie.201701439
(a) T. Akiyama, Chem. Rev. 107 (2007) 5744-5758;
(b) M. Terada, Synthesis 42 (2010) 1929-1982;
(c) D. Parmar, E. Sugiono, S. Raja, et al., Chem. Rev. 114 (2014) 9047-9153;
(d) I. Cori Čorić, B. List, Nature 483 (2012) 315-319;
(e) C. Zheng, S.L. You, Chem. Soc. Rev. 41 (2012) 2498-2518;
(f) D. Wang, D. Astruc, Chem. Rev. 115 (2015) 6621-6686.
(a) R. Zhao, G. Feng, X. Xin, et al., Chin. Chem. Lett. 30 (2019) 1432-1434;
(b) Z. Wang, Y. Mao, H. Guan, et al., Chin. Chem. Lett. 30 (2019) 1241-1243;
(c) P. Ye, X. Liu, G. Wang, L. Liu, Chin. Chem. Lett. 32 (2021) 1237-1240;
(d) H. Guan, L. Chen, L. Liu, Acta Chim. Sinica 76 (2018) 440-444.

Scheme 1 Overview of catalytic asymmetric synthesis of α-substituted 1, 3-dihydroisobenzofurans.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



