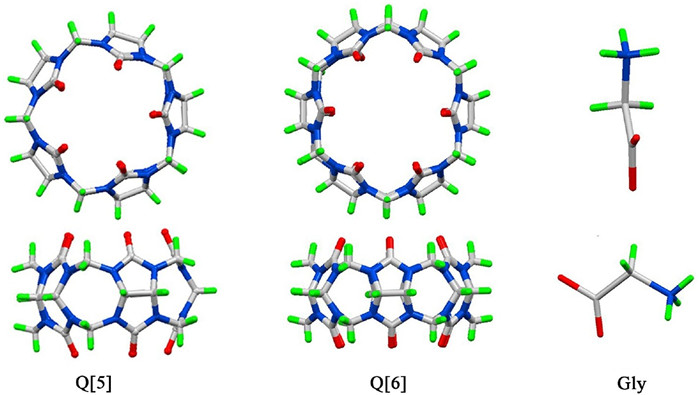
Figure 1.
Chemical structures of Q[5], Q[6] and Gly.
Recognition of glycine by cucurbit[5]uril and cucurbit[6]uril: A comparative study of exo- and endo-binding
Peihui Shan , Ruilian Lin , Ming Liu , Zhu Tao , Xin Xiao , Jingxin Liu

Recognition and determination of amino acids, which play fundamental and significant roles in life systems, are of great interest in pharmaceutical science and materials science for potential applications in nutritional analysis, medical diagnostics, cell imaging, and drug delivery [1]. Recently, several supramolecular systems that can recognize specific amino acids or their residues in peptides and proteins have been developed [2-4]. For example, Wei et al. successfully developed a fluorescent sensor based on a functionalized pillar [5]arene, which exhibits high selectivity and sensitivity towards L-tryptophan [4a]. Rao and co-workers synthesized a series of transition metal complexes with calix[4]arene derivatives, which demonstrate excellent recognition features toward some amino acids [5].
In the vein of developing supramolecular systems for recognition amino acids, our efforts have been focused on the cucurbit[n]uril (n=5-8, Q[n]), a family of synthetic macrocyclic cavitands made of glycoluril units linked by methylene bridges (Fig. 1) [6]. Over the past two decades, Q[n]s have been applied in numerous areas such as sewage treatment, metal ion separation, catalysis, electrochemistry and photochemistry. They also be used to recognize and bind amino acids, peptides and proteins through the combination of ion-dipole, hydrogen bonding interactions, van der Waals interactions as well as hydrophobic effects [7-18]. Most of these works have focused on the larger Q[n] homologues (Q[8] and Q[7]), because their spacious hydrophobic cavity can accommodate one or more amino acid residues to form host-guest complex. For example, Urbach group and Kim group have demonstrated that Q[8] and Q[7] have strong affinity for numerous peptides and proteins [7]. The N-teminal aromatic residues of peptides and proteins can be encapsulated within the cavities of the hosts Q[8] and Q[7]. By contrast, the smaller Q[n] homologues Q[5] and Q[6] have smaller hydrophobic cavity and smaller coordination portal, which makes them even more difficult to recognize and encapsulate amino acids. And naturally very little attention was devoted to the smaller Q[n] homologues, especially for the smallest Q[5].
Not long ago we and others have found that Q[6] and its derivative α, α', δ, δ'-tetramethyl-Q[6] can bind to some amino acids through exo-binding [16-18]. Inspired by these findings, we start to investigate the binding interactions of the amino acids with the Q[5] and Q[6]. In this paper, we report the extraordinary binding modes of the glycine (Gly), the smallest of the 20 nonessential amino acids, with the Q[5] and Q[6] in aqueous solution and in the solid state through 1H NMR spectroscopy and X-ray crystallography. Our objective was to reveal how the Q[5] and Q[6] recognize the Gly, and how the cavity size affects the possible binding modes.
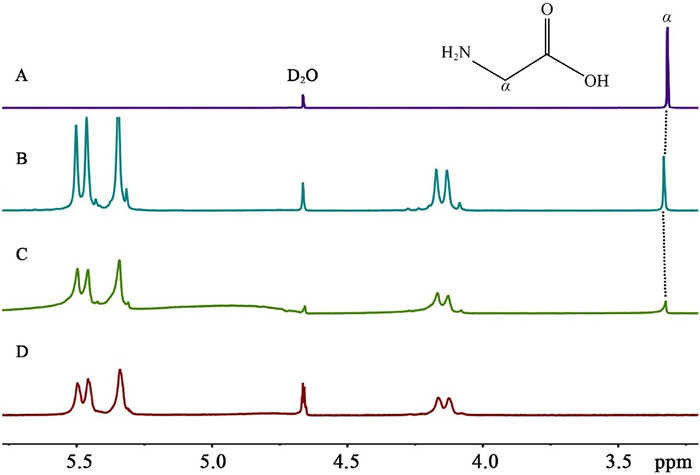
Exo- and endo-binding modes of the Gly with the hosts Q[5] and Q[6] in aqueous solution can be monitored by 1H NMR titration experiments. It is well known that the hydrophobic cavity of Q[n] is proton-shielding region which would induce upfield shift of the corresponding proton signals, while the outside of the portals of Q[n] belong to proton-deshielding region which would induce downfield shift of the corresponding proton signals [19, 20]. On this theory, it is easy for us to deduce the relative position of the guest and the host from their 1H NMR spectra. As can be seen in Fig. 2, the 1H NMR spectrum of Gly in D2O exhibits only one resonance signal of the methylene protons. In the presence of different equivalents of Q[5] host, the methylene proton signal of Gly slightly shifted downfield (from 3.276 ppm to 3.306 ppm), indicating that Gly is located at the deshielding region of the Q[5]. In other words, the Gly is located near the Q[5] portals. On the basis of this observation, and combined with the ESI-MS of Gly with Q[5] (Fig. 3) which showed the charged peak at 981.0145 for {Gly∩Q[5]∩Gly·H}+ (calcd. 981. 3167), we conclude that Gly can bind exo to Q[5] to form exclusion complex Gly∩Q[5]∩Gly.

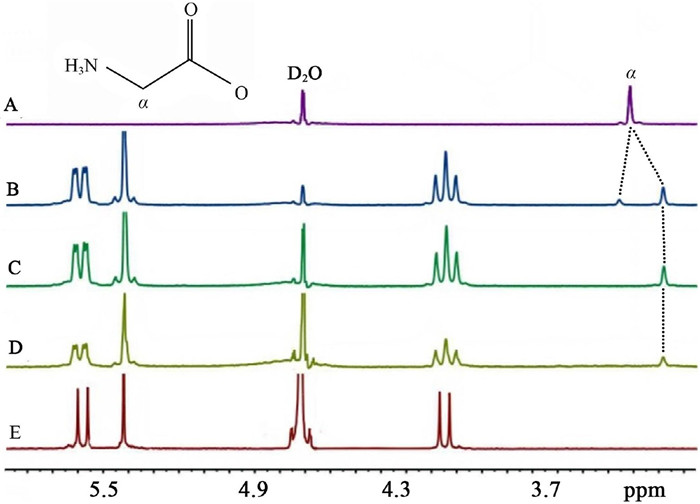
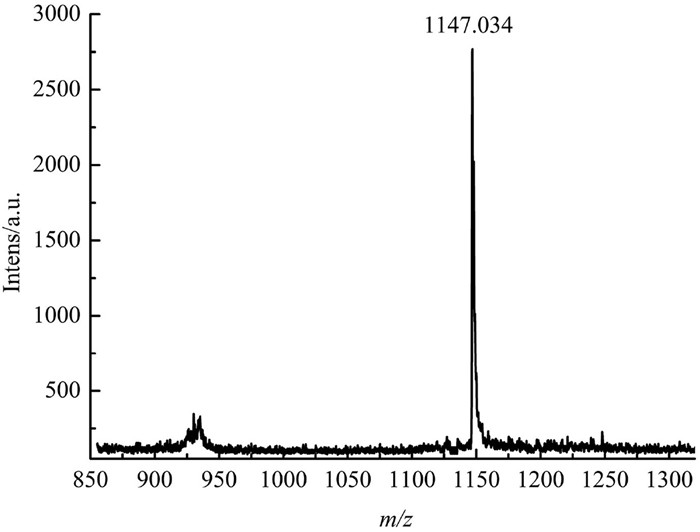
In contrast to Q[5] host, studies of the binding behavior of the Gly with the bigger Q[6] revealed completely different results. The 1H NMR spectra of Gly and Q[6] are shown in Figs. 4A and E, respectively. Upon the addition of 0.63 equiv. of Q[6] to the aqueous solution of Gly, two sets of signals of Gly were observed. One is on the upfield and the other is on the downfield (Fig. 4B). This observation suggests that the Gly reside inside and outside of the Q[6] host simultaneously in the presence of small amount of Q[6]. Obviously, Gly exhibits two binding modes (endo- and exo-binding) with the Q[6], generating an inclusion-exclusion hybrid complex Gly@Q[6]∩Gly. Notably, though, there are also two different types of complexes, inclusion complex Gly@Q[6] and exclusion complex Gly∩Q[6] in theory. The presence of the inclusion-exclusion hybrid complex Gly@Q[6]∩Gly is further evidenced by ESI-MS (Fig. 5). The peak at 1147.034 can be assigned to Gly@Q[6]∩Gly (calcd. {Gly@Q[6]∩Gly·H}+, 1147.37). When more and more Q[6] was added, the proton signals in the downfield disppeared (Fig. 4C and D), and only one set of proton signals of the Gly on the upfield are observed. It is clear that all the Gly molecules were encapsulated into the hydrophobic cavity of Q[6] host. It also suggests that the Gly molecules bind endo with the Q[6], forming only the inclusion complex Gly@Q[6] in the presence of enough of Q[6] host.
Above described 1H NMR experiments confirm that the Gly molecule can only bind exo to Q[5] to form exclusion complex because the Q[5] cavity is not large enough to accommodate the Gly molecule. For the Q[6] host, however, the binding modes depend on the quantities of the host. In the presence of enough of Q[6] host, the Gly molecule prefers to be encapsulated into the Q[6] cavity, and binds endo to Q[6] portals to form inclusion complex. In the presence of small amount of Q[6] host, the excessive Gly molecule also binds exo with the Q[6] to form exclusion complex.
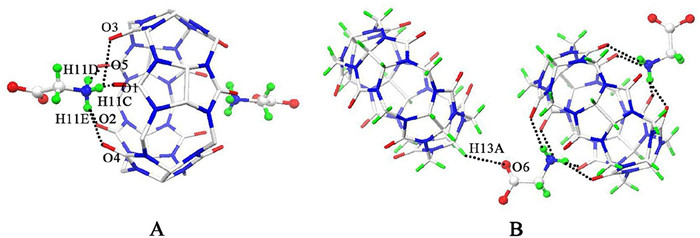
In order to get the direct information of the binding interactions of the Gly with the Q[5] and Q[6], we also try to obtain their single-crystals. Slow vapor evaporation of the aqueous solution of Gly with Q[5] results in the single-crystals of compound 1, which crystallized in the tetragonal crystal system, space group I41/a (No. 88). X-ray structural analysis reveals that the asymmetric unit of the compound 1 contains one half of Q[5] host, one Gly molecule, and five lattice water molecules. As shown in Fig. 6, the Gly molecule is located outside of the portals of the Q[5] host, which is in agreement with the 1H NMR spectroscopic data. The amine group of the Gly connects with the carbonyl oxygen atoms (O1, O2 and O5) of the Q[5] host through three hydrogen bondings with N–H–O distances of 1.971, 2.075 and 2.082 Å, respectively (Fig. 6A). In other words, two Gly molecules bind exo with the Q[5] host, generating an exclusion complex of Gly∩Q[5]∩Gly with 2:1 stoichiometry, which is consistent with their NMR results. At the same time, the carboxyl group on the other side of the Gly is interacts with the CH group of the neighbouring Q[5] host with C–H···O distance of 2.560 Å (Fig. 6B). Obviously, hydrogen bonding between ammonium groups of Gly and portal oxygen atoms of Q[5] accompanied by the ion-dipole interaction between N-terminal nitrogen and carbonyl oxygen atoms are the important driving forces for the exclusion complex formation.
Homoternary inclusion complex in which the Q[6] cavity accommodated two Gly molecules. The compound 2 was prepared in the presence of CdCl2. The X-ray crystal structure of compound 2 reveals a unique endo-binding structure, which crystallized in the cubic system with the Ia 3 (no. 205) space group. The asymmetric unit of the compound 2 consists of one half of Q[6] host, one protonated Gly molecule, one tetrahedral [CdCl4]2- anion, and eleven lattice water molecules. As can be seen in Fig. 7, the carboxyl group of the Gly is located inside the Q[6] cavity while the amine group of the Gly remain outside of the portal. A closer observation reveals that the carbon atom C19 of the Gly is 0.287 Å above the mean plane of the six carbonyl oxygen atoms of the Q[6] portal, indicating that the methylene resides outside of the Q[6] portal, which is distinctly different from what we observed in the aqueous solution. The amine group of the Gly also forms two hydrogen bondings with the carbonyl oxygen atoms (O5 and O6) of the Q[6] host with N–H–O distances of 2.083 and 2.165 Å respectively. On the whole, two Gly molecules bind endo with the Q[6] host, generating a homoternary inclusion complex Gly2@Q[6].
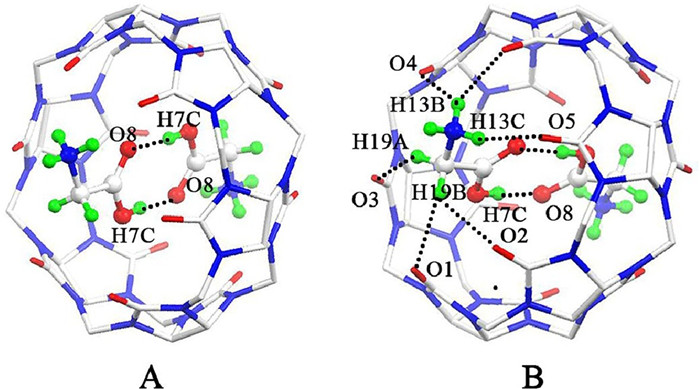
The most remarkable feature of the compound 2 is that two carboxyl groups of two encapsulated Gly molecules are connected to each other through hydrogen bonding with O–H···O distance of 1.729 Å. It is well known that the spacious hydrophobic cavity of Q[8] can simultaneously accommodate two guests. More often than not, charge-transfer complex formation was observed inside the hydrophobic cavity [21]. However, in the case of the inclusion complex Gly2@Q[6], two Gly molecules were observed be encapsulated into the small hydrophobic cavity of Q[6]. Except for the well known hydrophobic effects [22], the main reasons which contribute to the formation of this homoternary inclusion complex include: (i) O–H···O hydrogen bondings between two carboxyl groups of two encapsulated Gly molecules, (ii) N–H···O hydrogen bondings between the amine group of the Gly and the carbonyl oxygen atoms on the Q[6] portal, (iii) ion-dipole interactions between the quaternized nitrogen of the Gly and the carbonyl oxygen atoms on the Q[6] portal, as well as (iv) van der Waals interactions between the surfaces of the Gly and the inner wall of the Q[6].
In summary, this study explored the recognition feature of the hosts Q[5] and Q[6] toward the Gly molecule in aqueous solution and solid state. The binding interaction of the hosts Q[5] and Q[6] with the Gly reveals two different binding modes (exo and endo binding), depending on the size of the hydrophobic cavity of the hosts. Experimental data indicate that the Gly binds exo with the Q[5] host, forming exclusion complex Gly∩Q[5]. However, the Gly can binds endo and exo with the Q[6] host, generating hybrid complex Gly@Q[6]∩Gly in aqueous solution and inclusion complex Gly2@Q[6] in solid state. Interestingly, hydrogen bondings between two encapsulated Gly molecules were observed in the inclusion complex Gly2@Q[6] in solid state. The use of the smaller Q[n] homologues Q[5] and Q[6] to recognize Gly will greatly extend the application of Q[n]s.
The authors declare no conflict of interest.
This work was supported by the National Natural Science Foundation of China (No. 21861011), the Innovation Program for High-level Talents of Guizhou Province (No. 2016-5657), the Major Program for Creative Research Groups of Guizhou Provincial Education Department (2017-028), the Science and Technology Fund of Guizhou Province (No. 2018-5781) and the Natural Science Foundation of Anhui Province of China (Nos. 1808085MB43, 2008085MB36).
(a) M. Freidman, J. Agric. Food Chem. 47 (1999) 3457-3479;
(b) Y. Zhou, J. Yoon, Chem. Soc. Rev. 41 (2012) 52-67;
(c) H. Heli, M. Hajjizadeh, A. Jabbari, A. Moosavi-Movahedi, Anal. Biochem. 388 (2009) 81-90;
(d) Q.P. Duan, Y. Cao, Y. Li, et al., J. Am. Chem. Soc. 135 (2013) 10542-10549;
(e) B.B. Shi, K.C. Jie, Y.J. Zhou, et al., J. Am. Chem. Soc. 138 (2016) 80-83;
(f) G.C. Yu, G.P. Tang, F.H. Huang, J. Mater. Chem. C 2 (2014) 6609-6617;
(g) G.C. Yu, D. Wu, Y. Li, et al., Chem. Sci. 7 (2016) 3017-3024;
(h) Y. Cao, X.Y. Hu, Y. Li, et al., J. Am. Chem. Soc. 136 (2014) 10762-10769;
(i) M. Liu, J.L. Kan, Y.Q. Yao, et al., Sensors Actuators B: Chem. 283 (2019) 290-297;
(j) R.H. Gao, L.X. Chen, K. Chen, Z. Tao, X. Xiao, Coord. Chem. Rev. 348 (2017) 1–24.
(a) C.B. Lebrilla, Acc. Chem. Res. 34 (2001) 653-661;
(b) W. Si, P. Xin, Z.T. Li, J.L. Hou, Acc. Chem. Res. 48 (2015) 1612–1619;
(c) A. Casnati, F. Sansone, R. Ungaro, Acc. Chem. Res. 36 (2003) 246-254;
(d) K. Yang, Y. Pei, J. Wen, Z. Pei, Chem. Commun. 52 (2016) 9316–9326;
(e) M. Giuliani, I. Morbioli, F. Sansone, A. Casnati, Chem. Commun. 51 (2015)14140–14159;
(f) R. Pinalli, A. Pedrini, E. Dalcanale, Chem. Soc. Rev. 47 (2018) 7006-7026.
(a) A.R. Urbach, V. Isr. Ramalingam, J. Chem. 51 (2011) 664;
(b) A.T. Wright, M.J. Griffin, Z.L. Zhong, et al., Angew. Chem. Int. Ed. 44 (2005) 6375-6378;
(c) Q.H. Bai, S.W. Zhang, H.R. Chen, et al., ChemistrySelect 2 (2017) 2569-2573.
(a) T.B. Wei, J.F. Chen, X.B. Cheng, et al., Org. Chem. Front. 4 (2017) 210-213;
(b) L. Chen, W. Si, L. Zhang, et al., J. Am. Chem. Soc. 135 (2013) 2152-2155;
(c) S. Tashiro, M. Tominaga, M. Kawano, et al., J. Am. Chem. Soc. 127 (2005) 4546-4547;
(d) P. Wang, Z.T. Li, X.F. Ji, Chem. Commun. 50 (2014) 13114-13116;
(e) C.J. Li, J.W. Ma, L. Zhao, et al., Chem. Commun. 49 (2013) 1924-1926;
(f) B. Hua, J. Zhou, G.C. Yu, Tetrahedron Lett. 56 (2015) 986-989.
(a) J.P. Chinta, A. Acharya, A. Kumar, C.P. Rao, J. Phys. Chem. B 113 (2009) 12075-12083;
(b) R.K. Pathak, J. Dessingou, C.P. Rao, Anal. Chem. 84 (2012) 8294-8300;
(c) R.K. Pathak, V.K. Hinge, K. Mahesh, et al., Anal. Chem. 84 (2012) 6907-6913;
(d) J. Dessingou, A. Mitra, K. Tabbasum, G.S. Baghel, C.P. Rao, J. Org. Chem. 77 (2012) 371-378;
(e) R. Joseph, B. Ramanujam, A. Acharya, C.P. Rao, J. Org. Chem. 74 (2009) 8181-8190.
(a) J. Lagona, P. Mukhopadhyay, S. Chakrabarti, L. Isaacs, Angew. Chem. Int. Ed. 44 (2005) 4844-4870;
(b) X.L. Ni, X. Xiao, H. Cong, et al., Chem. Soc. Rev. 42 (2013) 9480-9508;
(c) E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah, X. Lu, RSC Adv. 2 (2012) 1213-1247;
(d) K.I. Assaf, W.M. Nau, Chem. Soc. Rev. 44 (2015) 394-418;
(e) S.J. Barrow, S. Kasera, M.J. Rowland, J. Barrio, O.A. Scherman, Chem. Rev. 115 (2015) 12320-12406;
(f) X.L. Ni, S.Y. Chen, Y.P. Yang, Z. Tao, J. Am. Chem. Soc. 138 (2016) 6177-6183.
(a) L.A. Logsdon, C.L. Schardon, V. Ramalingam, S.K. Kwee, A.R. Urbach, J. Am.
Chem. Soc. 133 (2011) 17087-17092;
(b) J.M. Chinai, A.B. Taylor, L.M. Ryno, et al., J. Am. Chem. Soc. 133 (2011) 8810-8813;
(c) L.A. Logsdon, A.R. Urbach, J. Am. Chem. Soc. 135 (2013) 11414-11416;
(d) L.C. Smith, D.G. Leach, B.E. Blaylock, O.A. Ali, A.R. Urbach, J. Am. Chem. Soc. 137 (2015) 3663-3669.
(a) M.V. Rekharsky, H. Yamamura, Y.H. Ko, et al., Chem. Commun. (2008) 2236-2238;
(b) W.J. Kim, K. Kim, H.I. Kim, Angew. Chem. Int. Ed. 126 (2014) 7591-7595;
(c) J.W. Lee, H.H.L. Lee, Y.H. Ko, K. Kim, H.I. Kim, J. Phys. Chem. B 119 (2015) 4628-4636;
(d) L.T. Li, M. Liu, L.D. Yue, et al., Anal. Chem. 92 (2020) 9322-9329;
(e) L.D. Yue, C. Sun, Q. Cheng, et al., Chem. Commun. 55 (2019) 13506-13509;
(f) S. Combes, K.T. Tran, M.M. Ayhan, et al., J. Am. Chem. Soc. 141 (2019) 5897-5907.
(a) A. Hennig, H. Bakirci, W.M. Nau, Nat. Methods 4 (2007) 629-632;
(b) D.M. Bailey, A. Hennig, V.D. Uzunova, W.M. Nau, Chem. Eur. J. 14 (2008) 6069-6077;
(c) A. Praetorius, D.M. Bailey, T. Schwarzlose, W.M. Nau, Org. Lett. 10 (2008) 4089-4092;
(d) W.M. Nau, G. Ghale, A. Hennig, H. Bakirci, D.M. Bailey, J. Am. Chem. Soc. 131 (2009) 11558-11570;
(e) H. Yin, X.J. Zhang, J.W. Wei, et al., Theranostics 11 (2021) 1513-1526;
(f) H. Yin, D. Bardelang, R.B. Wang, Trends Analyt. Chem. 3 (2021) 1-4.
M. Pozo, P. Hernàndez, L. Hernàndez, C.J. Quintana, Mater. Chem. 21 (2011) 13657–13663.
(a) S. Sonzini, T.J.S. Ryan, O.A. Scherman, Chem. Commun. 49 (2013) 8779-8781;
(b) K.I. Kuok, S.K. Li, Ian W. Wyman, R.B. Wang, Ann. N.Y. Acad. Sci. 1398 (2017) 108-119.
M.A. Gamal-Eldin, D.H. Macartney, Org. Biomol. Chem. 11 (2013) 488-495. doi: 10.1039/C2OB27007B
T. Minami, N.A. Esipenko, B. Zhang, L. Isaacs, P. Anzenbacher Jr, Chem. Commun. 50 (2014) 61–63. doi: 10.1039/C3CC47416J
Y. He, Y. Liang, H. Yu, ACS Comb. Sci. 17 (2015) 409-412. doi: 10.1021/acscombsci.5b00045
(a) D.T. Dang, H.D. Nguyen, M. Merkx, L. Brunsveld, Angew. Chem. Int. Ed. 52 (2013) 2915-2919;
(b) H.D. Nguyen, D.T. Dang, J.L.J. van Dongen, L. Brunsveld, Angew. Chem. Int. Ed. 49 (2010) 895-898.
O. Danylyuk, V.P. Fedin, Cryst. Growth Des. 12 (2012) 550-555. doi: 10.1021/cg2013914
(a) P. Thuéry, Inorg. Chem. 50 (2011) 10558-10560;
(b) P. Thuéry, CrystEngComm 14 (2012) 8128-8136.
(a) J.M. Yi, Y.Q. Zhang, H. Cong, S.F. Xue, Z. Tao, J. Mol. Struct. 933 (2009) 112-117;
(b) P.H. Shan, S.C. Tu, R.L. Lin, et al., CrystEngComm 19 (2017) 2168-2171;
(c) T. Yin, S. Zhang, M.X. Li, C. Redshaw, X.L. Ni, Sens. Actuator. 281 (2019) 568-573.
(a) K. Moon, A.E. Kaifer, Org. Lett. 6 (2004) 185;
(b) A.E. Kaifer, Acc. Chem. Res. 47 (2014) 2160-2167;
(c) R.L. Lin, J.Q. Li, J.X. Liu, A.E. Kaifer, J. Org. Chem. 80 (2015) 10505-10511.
(a) Y.X. Qu, R.L. Lin, Y.Q. Zhang, et al., Org. Chem. Front. 4 (2017) 1799-1805;
(b) R.L. Lin, G.S. Fang, W.Q. Sun, J.X. Liu, Sci. Rep. 6 (2016);
(c) R.L. Lin, Y.P. Dong, Y.F. Hu, et al., RSC Adv. 2 (2012) 7754-7758.
(a) Y.H. Ko, E. Kim, I. Hwang, K. Kim, Chem. Commun. (2007) 1305-1315;
(b) Y. Liu, Y. Yu, J. Gao, Z. Wang, X. Zhang, Angew. Chem. Int. Ed. 49 (2010) 6576-6579;
(c) F. Biedermann, O.A. Scherman, J. Phys. Chem. B 116 (2012) 2842-2849;
(d) Z. Ji, J. Li, G. Chen, M. Jiang, ACS Macro Lett. 5 (2016) 588-592.
(a) F. Biedermann, V.D. Uzunova, O.A. Scherman, W.M. Nau, A.D. Simone, J. Am.
Chem. Soc. 134 (2012) 15318-15323;
(b) F. Biedermann, M. Vendruscolo, O.A. Scherman, A.D. Simone, W.M. Nau, J.
Am. Chem. Soc. 135 (2013) 14879-14888.

Figure 2 1H NMR spectra (400 MHz, D2O) of Gly (0.01 mol/L) in the absence (A) and in the presence of (B) 0.55 and (C) 1.30 equivalence of Q[5] in D2O at 20 ℃; (D) 1H NMR spectrum (400 MHz) of Q[5] in D2O (0.01 mol/L) at 20 ℃.

Figure 3 ESI-MS spectrum of Gly with Q[5]. The peak found atm/z 981.0145 corresponds to {Gly∩Q[5]∩Gly·H}+ (calcd. 981. 3167).

Figure 4 1H NMR spectra (400 MHz, D2O) of Gly (0.01 mol/L) in the absence (A) and in the presence of (B) 0.63, (C) 1.01 and (D) 2.10 equiv. of Q[6] in D2O at 20 ℃; (E) 1H NMR spectrum (400 MHz) of Q[6] in D2O (0.01 mol/L) at 20 ℃.

Figure 5 ESI-MS spectrum of Gly with Q[6]. The peak found at m/z 1147.034 corresponds to {Gly@Q[6]∩Gly·H}+ (calcd. 1147.37).

Figure 6 Ball-and-stick representation of compound 1 showing Gly molecules connect to portals (A) and outer surfaces (B) of the Q[5] host through hydrogen bonds, forming an exclusion complex of Gly∩Q[5]∩Gly. Solvate water molecules and chloride anions are omitted for clarity. O = red, C = grey and N = blue.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
