Citation:
Zhou Dawei, Li Chunping, Yin Furong, Tang Xin, Pu Chunying, He Chaozheng. Two-dimensional 1T-PS2 as a promising anode material for sodium-ion batteries with ultra-high capacity, low average voltage and appropriate mobility[J]. Chinese Chemical Letters,
2020, 31(9): 2325-2329.
doi:
10.1016/j.cclet.2020.04.045
Two-dimensional 1T-PS2 as a promising anode material for sodium-ion batteries with ultra-high capacity, low average voltage and appropriate mobility
English
Two-dimensional 1T-PS2 as a promising anode material for sodium-ion batteries with ultra-high capacity, low average voltage and appropriate mobility
College of Physics and Electronic Engineering, Nanyang Normal University, Nanyang 473061, China
b.
Fundamental Department, Aviation University of Air Force, Changchun 130022, China
c.
Institute of Environmental and Energy Catalysis, School of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an 710021, China
d.
College of Material Science and Engineering, Guilin University of Technology, Guilin 541004, China
Received Date:
27 March 2020 Accepted Date:
23 April 2020 Revised Date:
20 April 2020 Available Online:
15 September 2020
Abstract:
As electrodes, two-dimensional materials show special advantages including the infinite planar lengths, broad electrochemical window, large surface-volume ratio, and much exposed active sites. In theory, the two-dimensional materials consist of the elements with high electronegativity may absorb more Na atoms, resulting in a high battery storage capacity. Based on the above idea, we selected the two dimensional metallic PS2 with 1T-Type structure as an anode material, and explored its potential applications as an electrode material for Na-ion battery through first-principle calculations. As we expected, when two dimensional PS2 is used as an anode in Na-ion battery, it can adsorb maximum three layers of sodium atoms on both sides of the monolayer, resulting in a maximum theoretical capacity of 1692 mAh/g. Furthermore, it also possesses a rather small sodium diffusion barrier of 0.17 eV, a low average open-circuit voltage of 0.18 V, and a relatively small lattice changes within 13% during the intercalation of Na. These results suggested that the two dimensional PS2 is a potentially excellent Na-ion battery anode. Our idea of designing two-dimensional anode materials with high storage capacity may provide some references for designing the next generation anode materials of metal-ion batteries.
As clean energy storage technologies, Li-ion batteries (LIBs) have achieved great success in the past ten years due to their many advantages such as high energy density, structural flexibility and stability, environmentally friendly [1-7]. However, the lack of lithium resources and the comparatively low battery storage capacity hinders its large-scale applications in the further [8]. To meet the demand for the next generation metal-ion batteries, great efforts have been carried out to develop new metal-ion batteries [9-17]. Among various anode material candidates for metal-ion batteries, two dimensional (2D) anode materials attract increasing attention, especially for sodium-ion batteries (SIBs) [12, 10-17]. Firstly, compared with lithium, sodium is more abundant and safe. Secondly, different from their bulk materials, 2D materials have unique chemical and physical characterization, which show great potentials for various applications such as field-effect transistors [18], p-n junctions [19], sensing materials [20, 21], energy storage and conversion [22, 23]. Especially, benefiting from a large surface-area-to-volume ratio, 2D materials have a larger contact area between the electrolyte and electrode, which is usually favorable for the improvement of energy density. Hence 2D SIBs are believed to be promising candidates, which can solve the problems of LIBs. Unfortunately, the anode materials with good-performance in well-developed LIBs usually do not apply to SIBs. The atomic radius of Na is larger than that of Li, which usually leads to large volume strain during charge/discharge, low reversible capacity and poor cycling stability. Therefore, designing and preparation of new 2D SIBs anode materials with good performance are very necessary.
Theoretical investigations based on first-principle calculation play a very important role in understanding the charge/discharge process and the resulting physical and chemical changes of electrode materials in metallic-ion battery. Up to known, a great deal of 2D materials, including graphene systems [24, 25], phosphorene [26, 27], MXene [28, 29], transition-metal dichalcogenides and nitrides [10, 30-34], have been discovered theoretically to show excellent performance in SIBs. For example, when used as anodes in SIBs, phosphorene reaches a theoretical capacity of 865 mAh/g [26], and borophene gains a maximum theoretical capacity of 1984 mAh/g [35]. Other 2D materials such as defective graphene (1450 mAh/g) [36], borocarbonitride based anode (810 mAh/g) [37], silicene (954 mAh/g) [38, 39], B-doped graphene (762 mAh/g) [40] and double-layer graphene-phosphorene hybrid (372 mAh/g) [41] were also investigated as potential anode materials for SIBs by means of first-principle calculations. The ion diffusion of those 2D materials are roughly ranges from 0.1 eV to 0.6 eV. However, searching 2D materials with good performance for applications in SIBs still faces great challenges such as volume expansion problem, capacity restrict and diffusion barrier issue.
Among these issues for the next generation metal-ion batteries, the low storage capacity of anodes is the most important and urgent problem. For example, the storage capacity of graphite is only 372 mAh/g for lithium battery [42, 43]. Moreover, when graphite is applied to sodium battery as an anode material, the storage capacity decreases dramatically to a very small value of 35 mAh/g [42, 43]. In theory, electronegativity is an important factor, which affects the electrochemical potential of electrodes. A large difference in electronegativity usually results in the formation of a more ionic bond. Strong electronegativity elements usually can gain more electron transfers from sodium atoms, forming a stronger ionic bond and absorbing more sodium atoms. So from the perspective of 2D anode material design, 2D materials composed of the elements with high electronegativity may gain large capacity storage in the metal-ion battery. For example, due to the higher electronegativity of nitrogen, nitrogen doping to carbon (N—C) could lead N—C with the enhanced lithium-ion storage properties [44, 45]. For the same reason, N and S doped in graphene also result in a reversible high capacity of 1090 mAh/g [46]. So in this paper, we selected 2D 1T-PS2 as a potential anode material and investigated its performances in sodium-ion battery. In fact, the solid PS2 with layered structure at zero pressure has been synthesized experimentally reported in a recent work [47]. Furthermore, the monolayer PS2 with 1T-type structure2 was also proposed in the previous paper [48]. Most importantly, 2D PS2 exhibits metallic character and is composed of comparatively highly electronegative elements P and S, so it meets our requirement for 2D anodes with high battery storage capacity. Expect for superconductivity property, the structural stability and some important physical properties of the 2D PS2 such as electronic structure and mechanical properties are also investigated systematically.
In this paper, we first checked the stability of PS2 monolayer through first-principle calculations, and then investigated its performance as an anode material for sodium-ion battery. As we expected, our calculations show that 2D PS2 is a superior anode with a maximum theoretical battery capacity of 1692 mAh/g. We further show that the other performances of 2D PS2 as an anode for SIBs such as the ion diffusion barrier and the open current voltage are also excellent.
The first-principles calculations were performed with the projector augmented wave (PAW) method [49, 50] as implemented in the Vienna ab initio simulation package (VASP) [51, 52]. The electron exchange-correlation energy was treated within the generalized gradient approximation (GGA), using the functional of Perdew, Burke, and Ernzerhof (PBE) [53]. The energy cutoff of the plane wave was set to 380 eV and the Brillouin zone was sampled with a 12×12×1 Monkhorst-Pack k-point grid. All the atomic positions were fully optimized with the convergence of 10-5 eV and 10-3 eV/Å for energy and force, respectively. To avoid the interactions between periodic images, a large vacuum space of 35 Å in the perpendicular direction of the sheet was used The finite displacement method as implemented in the phonopy package [54] was used to calculate the phonon dispersion curves. In order to determine the dynamical stability of the PS2 monolayer, the thermal stability was analyzed by ab initio molecular dynamics (AIMD) simulations using the canonical ensemble (NVT) with a 3×3×1 supercell. In the calculation of sodium ions diffusion, we used the nudged elastic band (NEB) method to get the ion diffusion/barrier [55].
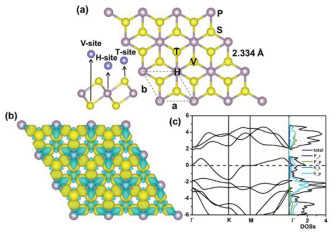
Monolayer PS2 adopts the AB2 structures [48] found in many two-dimensional transition metal disulfides, and we found that it is energetically more stable in its 1T phase rather than its 1H phase. As shown in Fig. 1a, the structure of PS2 consists of three atomic sub-planes similar to 1T-MoS2. The sub-plane of P atoms is sandwiched between the two sub-planes of sulfur atoms. The optimized lattice constants are a = b =3.288 Å with a layer thickness of 2.72 Å and the distance of P–S is 2.334 Å. To evaluate the chemical bonding nature, we computed the charge difference density as shown in Fig. 1b, which is defined as the total electron density of the PS2 monolayer minus the electron density of isolated P and S atoms at their respective positions. It is obviously seen that for the P–S bonds, the transfer charges are shifted toward S atoms, indicating the existence of polar covalent bonds between P–S atoms. According to the Bader charge population analysis, the charge transfer from P atom to S atom in PS2 monolayer is about 0.44 |e|, showing strong interactions between P and S atoms. We further calculated the electronic band structures and projected density of states. As shown in Fig. 1c, PS2 exhibits metallic character due to severe energy levels crossing the Fermi level. From the projected DOS analysis, we can see that the metallic states at the Fermi level are mainly contributed by S-2p states together with minor contributions from P-2s states. Hence, the 2D PS2 is formed by the mixture of metallic and polar covalent bonds. We also want to point out that most of the stable phases for 2D disulfides (e.g., MoS2, TiS2, CrS2) [11, 56] are semiconductors with poor conductivity, while the metallic 1T-type phases of which usually exist as metastable phases. The inherent metallic character of the stable 2D PS2 with 1T-type structure is favorable for its application as an anode material.
Figure 1
Figure 1.
(a) The structure of two-dimensional PS2 monolayer. P and S atoms are represented by gray and yellow spheres, respectively. (b) Difference charge density of PS2. The gold color (i.e., 0.005 e/Å3) in the plot indicates an electron density increase in the electron density after bonding, and the cyan color (i.e., 0.005 e/Å3) indicates a loss.
However, before we investigated the physical and chemical properties of 2D PS2 systematically, we first explored the structural stability of the PS2 monolayer. The binding energy is calculated, which is defined by the following formula:
(1)
where EP(ES) and EPS2 represents the total energies of a single P(S) atom and PS2 monolayer, respectively. The binding energy of the PS2 monolayer is 4.54 eV/atom, higher than that of silicene and germanene (3.98 and 3.26 eV/atom, respectively) [57], suggesting that the P–S bonds in PS2 monolayer are robust. While for 1H-PS2 phase, the binding energy is lower than that of 1T-PS2 phase (4.16 eV/atom), indicating that 1T-PS2 is stable energetically than 1H-PS2. So in this paper, we only focuses on 1T-PS2, and investigated it's performances as an anode material. Also, we estimated the dynamical stability of 2D PS2 monolayer according to its phonon curves. As shown in Fig. S1a (Supporting information), all vibrational modes were found to be real, confirming that PS2 monolayer is dynamically stable. The highest frequency of the optical mode is up to 17.24 THz (~575 cm-1), which was comparable to those of black phosphorene (~450 cm-1) [58] and MoS2 (~500 cm-1) [59], indicating the strong bonding characteristic in the PS2 monolayer. In addition, we can see that the phonon eigenvectors exhibit a strongly mixed character of P and S atoms. To further confirm the thermal stability of the PS2 under normal conditions, we carried out AIMD simulations in NVT, running for 10 ps at 600 K with a time step of 1 fs. The fluctuation of the total potential energy as a function of simulation time is shown in Fig. S1b (Supporting information), which shows that the average value of the total potential energy remains nearly constant during the entire simulation. The final structural configuration of 2D PS2 at the end of the molecular dynamical simulations is also illustrated in Fig. S1b, revealing that the structure does not experience serious structural disruptions. So the PS2 monolayer possesses good thermal stability and can maintain structural stability at a temperature of 600 K.
We also examined the mechanical stability of PS2 monolayer by calculating its linear elastic constants. The elastic constants of monolayer PS2 are C11 = C22 = 78 N/m, C12 = 45 N/m, and C66 = 16.5 N/m, respectively, which meet the necessary mechanical equilibrium conditions [60] for mechanical stability: C11C22 - C122 > 0 and C11, C22, C66>0, confirming the mechanical stability of PS2. We calculated the in-plane Young's modulus (Y) of PS2, which is defined as: Y=(C11 - C122)/C22. The Young's modulus of PS2 is about 52 N/m. It is worth noting that this value is lower than that of silicene (62 N/m) and TiS2 (74 N/m) [11, 61]. Therefore, the structure of PS2 shows better mechanical flexibility, which is favorable for the manufacture of flexible battery materials.
Since the 2D PS2 is expected to be an excellent anode material with a very large capacity for SIBs. We firstly investigated the adsorption of a single Na atom on the surface of PS2 monolayer by constructing a 2×2×1 supercell associated with the chemical stoichiometry of NaP4S8. The adsorption energy of Na is defined as:
(2)
where EPS2Na and EPS2 are the total energies of the Na adsorbed PS2 monolayer and pristine PS2 monolayer, respectively, μNa is the chemical potential of Na and is taken as the cohesive energy of bulk Na. The negative value of adsorption energy implies that the Na atom prefers to be adsorbed on the monolayer instead of forming a bulk metal. Considering the lattice symmetry of PS2 monolayer, three possible adsorption sites are considered, as shown in Fig. 1a. After our geometrical optimization, the adsorption energies of three sites are -0.89, -0.80 and -0.44 eV for V-, H- and T-sites, respectively. The adsorption energies of three sites are negative, implying that Na atom prefers to be adsorbed on the host materials rather than forming a cluster.
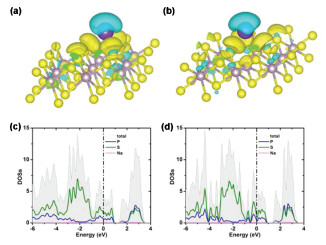
To further understand the adsorption of Na atom, we make the Bader charge analysis and the Na atoms possess a charge of 0.82 and 0.83 |e| at V- and H-sites, respectively, which means that the charge transfer from Na atom to adjacent sulfur atoms. The charge transference induced by Na atom suggests that the adsorption is chemical and hence can be seen as a redox reaction during the charge/discharge process. The existence of chemical adsorption also can be confirmed by the charge density difference (Figs. 2a and b), which is defined by
Figure 2
Figure 2.
The density of states of NaPS2 with Na atom at (a) V-site and (b) H-site. The charge density difference with the adsorption of Na atom at (c) V-site and (d) H-site. The gold color (i.e., 0.005 e/Å3) in the plot indicates an electron density increase and the cyan color (i.e., 0.005 e/Å3) indicates a loss.
The density of states of the PS2 after adsorption of Na atom with V- and H-sites are also calculated, as shown in Figs. 2c and d, respectively. The results show that the system still keeping metallic character, which are suitable for making electrode materials from the PS2 monolayer.
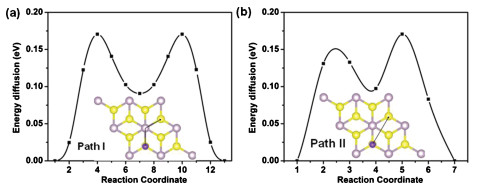
The energy barrier of Na-ion diffusion has an important effect on the charging and circuit rate capacity of SIBs. According to the above results, the data of adsorption energy shows a small difference of a Na ion at V- and H-sites (0.09 eV), which is due to the existence of a similar S environment at their adsorption sites. So the Na ions are expected to have a minimum energy path for Na diffusion from a V-site to the nearest neighboring V-site via the H-site (path Ⅰ). Furthermore, the path from V-site to adjacent V-site directly is taken into consideration (path Ⅱ). The paths and the relative energy profile are shown in Fig. 3, where the adsorption energy of Na atom at the V-site is taken as reference. The calculated diffusion barrier of Na ion along the path I is 0.17 eV, which is the lowest of two possible circumstances. It can be explained that this path can reduce the influence of the energy variation at different sites.
Figure 3
Figure 3.
Relative energy profile for the diffusion of Na ion on the surface of monolayer PS2 along path Ⅰ(a) and path Ⅱ(b). The inset is the top view of the trajectory of Na ion diffusion over the surface of PS2 monolayer.
The important parameters of SIBs as electrode materials are the open-circuit voltage (OVC) and theoretical storage capacity. The theoretical storage capacity is directly concerned with the number of adsorbed atoms. The intrinsic advantage of the monolayer materials is double Na storage capacity through adsorbing multilayer Na on both sides. The average adsorption energy layer by layer can be obtained by:
(4)
Here n represents the number of layers of Na atom, ENa8nP4S8 and ENa8(n-1)P4S8 are the total energy of PS2 with the adsorption of n and n-1 sodium atom layers, respectively. μNa is the chemical potential of Na. When a new layer of sodium atoms is added on both sides of 2D PS2, a negative Ea means that the adsorption of the new layer is stable energetically. The maximum storage capacity can be obtained by
(5)
where m is the number of adsorbed Na ions on the PS2 per formula unit, F (26, 801 mAh/g) is the Faraday constant, and MPS2 is the molar mass of PS2 per formula unit.
Three layers of Na atom on each side for Na ions adsorption on 2 × 2 × 1 supercell can be seen from Fig. 4a. The first Na atom layer is located at the V-site and the average adsorption energy is -0.45 eV. For the second layer, Na atoms prefer to be adsorbed at the T-site and the average adsorption energy becomes -0.06 eV. As for the third layer, Na atoms positions are the same as the first layer and the average adsorption energy is -0.02 eV. The third layer of Na adsorption weakens the adsorption ability of 2D PS2, but the adsorption energy remains negative. The comparatively low absorption energy implies a weak multilayered adsorption of Na atoms. However, the above adsorption energies of the second or third layer Na atoms are larger or comparable to than those of typical electrode materials, such as Ca2N (-0.003 eV per atom) [62] and GeS (-0.02 eV/atom) [63]. We also want to point out that the predicted theoretical capacity storage is usually larger than that of experimental results. Nonetheless, we give the maximum adsorption of three-layer Na atoms in theory, and show qualitatively that 2D PS2 is a potential anode with high capacity. The adsorption of three-layer Na atoms on each side of PS2 monolayer also can be understood by the distribution of the dispersive electron cloud acting as S ions, which are demonstrated by the electron local function (ELF) [64] as shown in Fig. 4b. The value of ELF is between 0.0 and 1.0. The values of 0.5 and 1.0 refer to fully delocalized and localized electrons, respectively, while 0.0 represents a very low charge density. For the first layer of Na atoms, the electron clouds of Na atoms are transferred to the S atoms in the 2D PS2, indicating strong attractions for Na atoms. For the second layer of Na atoms, the electron clouds can be seen clearly to shift toward the PS2 monolayer, showing the effect of attractions from 2D PS2. Even for the third layer of Na atoms, some degrees of electron cloud deviation from the centers of the absorbed Na atoms can be seen, revealing that the third layer of Na atoms also feels the attractions from 2D PS2. Both the electron cloud distributions as well as the negative adsorption energy indicate that the maximum three layers of Na atoms can be absorbed on the PS2 monolayer. The comparatively high electronegativities of P and S atoms enable 2D PS2 to absorb so many Na atoms, and finally gain a high battery capacity. The maximal theoretical capacity of the PS2 monolayer was calculated to be 1692 mAh/g, which is lower than the capacity of borophene (1984 mAh/g) [35], but is even larger than most other reported 2D disulfides (e.g., 146 mAh/g for MoS2 [32] 466 and 233 mAh/g for VS2 [65, 66], 479 mAh/g for TiS2 [11] and comparable to that of NiC3(-1698 mAh/g) [14]. During the Na ions intercalation process, the lattice constants in the x–y plane only experience a tensile strain of about 12.7%, which are comparable to the typical values that below 10% are acceptable.
Figure 4
Figure 4.
(a) The side view of the atomic structure of Na-intercalated PS2 monolayer, where three Na layers adsorbed on each side monolayer. (b) Electron localization function isosurface (0.5) of PS2 monolayer with three-layers of Na atom adsorbed on each side.
In addition, open-circuit voltage was computed to estimate the performance of the PS2 monolayer as an anode material. A low OVC of the anode implies the possibility of a high net cell voltage. The charge/discharge process of PS2 monolayer can be described as
For this reaction, when the change of volume and entropy during the adsorption process are neglected, the average open-circuit voltage can be defined by
(6)
If we define Eads=ENanPS2 - EPS2 - nENa, then we further get the following formula
(7)
where Eads is the adsorption energy before and after the adsorption of Na atoms, EPS2 and ENanPS2 are the total energies of the PS2 monolayer before and after the adsorption of Na atom, ENa is the energy per Na atom in its stable bulk structure of Na metal, n is the number of adsorbed Na content on a 3 × 3 × 1 supercell of PS2 monolayer. With the increase of the adsorbed Na concentration from 2 to 54 atoms on the 3 × 3 × 1 supercell, the OVC decreases from 0.94 V to about 0.18 V as shown in Fig. S2 (Supporting information). The dropping voltage with the increasing Na ion concentration has also been reported for other anode materials [11, 67]. In summary, PS2 possesses excellent stability and superior qualities and ultrahigh capacity for application as anode material in SIBs.
In conclusion, to design a 2D anode material with high battery capacity, we selected the 1T-type PS2 monolayer as a 2D anode material for Na-ion battery, which is composed of the comparatively high electronegative elements P and S. The structural stability of 2D PS2 was confirmed by dynamic, thermodynamic, and mechanical calculations. The 2D PS2 with 1T-type structure hosts excellent conductivity during Li/Na adsorption process. The strong electronegativities of P and S atoms in the 2D PS2 result in a maximum theoretical storage capacity of 1692 mAh/g, which is quite high among two-dimensional anode materials. Moreover, 2D PS2 possess a rather small sodium diffusion barrier of 0.17 eV, a low average open-circuit voltage of 0.18 V, and a relatively small lattice changes within 13% during the intercalation of Na. All these results suggest that the two dimensional PS2 is a promising anode material for sodium battery. In addition, our design concept of 2D anode materials with high battery storage capacity is reasonable, which may provide some references for the designing and improving metallic ions batteries in the further.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was supported by the Henan Joint Funds of the National Natural Science Foundation of China (Nos. U1904179 and U1404608, 21603109, U1404216), the National Natural Science Foundation of China (Nos. 21603109, 51501093) and the Key Science Fund of Educational Department of Henan Province of China (No. 20B140010).
Figure 1
(a) The structure of two-dimensional PS2 monolayer. P and S atoms are represented by gray and yellow spheres, respectively. (b) Difference charge density of PS2. The gold color (i.e., 0.005 e/Å3) in the plot indicates an electron density increase in the electron density after bonding, and the cyan color (i.e., 0.005 e/Å3) indicates a loss.
Figure 2
The density of states of NaPS2 with Na atom at (a) V-site and (b) H-site. The charge density difference with the adsorption of Na atom at (c) V-site and (d) H-site. The gold color (i.e., 0.005 e/Å3) in the plot indicates an electron density increase and the cyan color (i.e., 0.005 e/Å3) indicates a loss.
Figure 3
Relative energy profile for the diffusion of Na ion on the surface of monolayer PS2 along path Ⅰ(a) and path Ⅱ(b). The inset is the top view of the trajectory of Na ion diffusion over the surface of PS2 monolayer.
Figure 4
(a) The side view of the atomic structure of Na-intercalated PS2 monolayer, where three Na layers adsorbed on each side monolayer. (b) Electron localization function isosurface (0.5) of PS2 monolayer with three-layers of Na atom adsorbed on each side.

 DownLoad:
DownLoad:










 下载:
下载:





