Figure 1.
Three categories of well-established therapeutic agents.

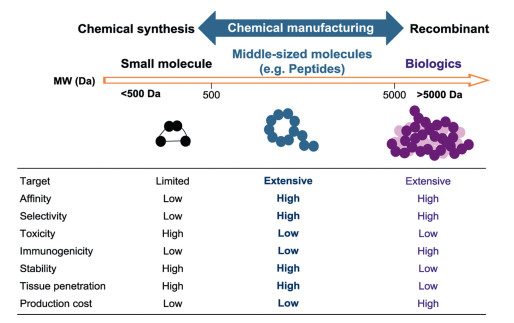
According to the molecular weights, therapeutic agents can be classified into three categories: biologics (>5000 Da), small molecules (< 500 Da), and middle-sized molecules (500-5000 Da) (Fig. 1). Biologics drugs, which are mostly composed of natural constituents, show the promising properties in regard with high target selectivity, high potency, and low toxicity. However, biologics usually exhibit low bioavailability, low metabolic stability and poor tissue permeability. Besides, most biologics are immunogenic, and the production costs of the biologics as therapeutic agents commonly are high. Compared to biologics, small-molecule drugs show low production cost, high oral bioavailability, high tissue permeability and high metabolic stability. Nonetheless, small-molecule drugs usually have reduced target selectivity, which may cause difficult-to-overcome medication side effects. The middle-sized molecules have molecular weights between biologics and small molecules, and they usually include peptides, nucleic acids and other related bioconjugates. Among them, peptides are attracting increasing attention as promising therapeutics because peptides possess the high affinity and selectivity towards targets, as well as the high tissue penetration [1-3]. Furthermore, peptides are more accessible to prepare by chemical methods than that of protein biologics. Chemical synthesis enables custom-made peptides to improve their pharmacokinetic properties [3-6].
New technologies such as constructing biological and synthetic peptide libraries, high-throughput screening or selection, and innovative drug administration ways have paved the way for the increasing interest in therapeutically active peptides. Currently, hundreds of peptide therapeutics, which are almost exclusively composed of L-amino acids (L-peptides), are being developed. More than 150 are currently undergoing preclinical studies or in clinical trials, and over 60 peptides have been approved [7]. Peptides have been used as drugs (e.g., Insulin, Liragluride, Enfuvirtide and Etelcalcetide) for diabetes, anti-HIV, Alzheimer's disease, malaria, inflammation, pain, and antimicrobials (Fig. 2).

The development of peptide drugs sometimes can be severely hampered by their short half-life, limited administration routes, and latent immunogenicity when they are used for in vivo applications. Nonetheless, one interesting and promising strategy is to use D-peptides composed of D-amino acids. D-Peptides combine advantages of biologics and small molecules. First, D-peptides are fully resistant to proteases, which significant increases the serum half-life of peptides. Second, D-peptides are excellent oral drug candidates. Third, D-peptides have long shelf-lives and can be easily modified during the chemical synthesis. Fourth, D-peptides arenonor less immunogenic. Thus, D-peptides represent a new class of therapy and a series of D-peptide identification techniques have been developed to discover D-peptide ligands aslead compoundsfor the development of pharmaceuticals [8, 9].
Initially, retro-inverso isomerization strategy was used to develop D-peptide ligands, such as retro-inverted D-peptide (carvc) targeting the epidermal growth factor receptor pathway [10], ERGbound RI-EIP1 and RI-EIP2 [11]. However, the retro-inverso strategy only works on short peptides and usually does not lead to ligands with activity [12, 13]. To overcome the problem, alternative strategies, i.e., mirror-image phage display, mirror-image one-bead one-compound (OBOC), and virtual mirror-image approaches were developed. This review described the current state of D-peptide drug discovery by using the new highthroughput screening methods. We emphasized the critical role of protein chemical synthesis in D-peptide drug discovery. Besides, the delivery of D-peptides across cell membrane was also briefly discussed.
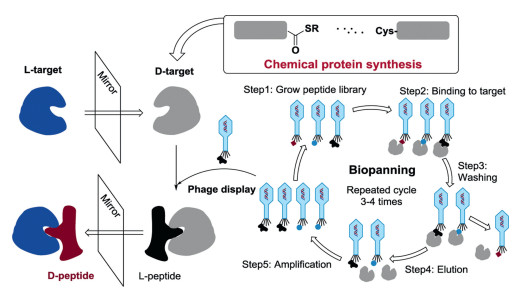
Mirror-image phage display is one of the most popular techniques used to discover the potentially bioactive D-peptides. Mirror-image phage display relies on a basic phenomenon that the structure of D-protein is the exact mirror-image enantiomer of the corresponding L-protein. This technology is an advanced version of the normal phage display approach allowing the selection of D-peptides. In a routine phage display method [14], a given L-target protein was directly used to select for L-peptides presented on the surface of phages and the verified L-peptides could be directly bind to the L-target protein. In the mirror-image phage display, a D-target protein, the mirror-image of the target L-protein, was first chemically synthesized and then employed for the phage display selection. The screening gave L-peptide ligands to bind the D-target proteins. Then, the selected L-peptides were translated into their mirror-image forms, D-peptides. These D-peptide ligands were expected to bind the L-target protein (Fig. 3).
Mirror-image phage display takes the advantage of the phage display, enabling the rapid and straightforward selection of peptide ligands from an encode peptide library with a huge structural diversity up to 1013. The mirror-image phage display can be even carried out without any structural information about the target protein. More importantly, D-peptides are resistant to proteolytic degradation and considered to be more suitable for diagnostic and therapeutic reagents [3].
Mirror-image phage display was first reported in 1996 to give the D-peptide ligands for SH3 domain of Src kinase [15]. To achieve this, the mirror-image protein (D-SH3) of the c-Src SH3 domain was synthesized by SPPS. Then, a 10-residue random L-peptide library was constructed and presented on the phage surface. After that, the phage display screening was performed to give several L-peptides that can selectively bind with D-SH3. Finally, the mirror-image peptides (D-peptides) of L-peptides would generate the binding affinity to the native c-Src SH3 domain. One of the D-peptides (denoted Pep-D1, rclsglrlglvpca, single-letter-coded D-amino acid residues in lower-case letters) could bound to SH3 with a dissociation constant (Kd = 63 μmol/L).
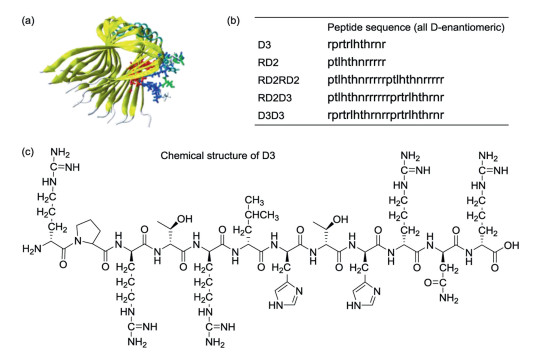
The discovery of D-peptide ligands for Alzheimer's disease is another important example to show the application of mirror-image phage display. Initially, Cribbs et al. demonstrated that the synthetic D-Aβ could be obtained with similar biological properties to L-Aβ [16]. The mirror-image phage display strategy was used for the selection of D-peptides from a huge randomized 12-mer peptide library to bind Aβ(1-42) [17]. Then, Willbold and colleagues identified that a dodecamer D-peptide D3 (rprtrlhthrnr) could bound to Aβ fibrils by using the mirror-image phage display technique [18, 19]. D3 can also strongly bind towards N-terminally truncated and pyroglutamated Aβ (pEAβ(3–42)) in vitro and in vivo in the new mouse model TBA2.1. Oral administration of D3 improved the behaviour in the Morris water maze [20].
The following studies showed D3 can improve the cognitive performance of AD transgenic mice by precipitating toxic Aβ species and converting them into large, nonfibrillar, and nontoxic aggregates [21]. Furthermore, D3 was labelled for mechanism study. In order to visualize amyloid in Alzheimer's disease, 125I-labeled and 18F-labeled D3 were prepared and tested in post mortem human autoradiography experiments. The 18F-labeled probes showed more promising visualization capacity of amyloid than the 125I-labeled probes. In the case of each 18F radio ligands, the grey matter uptake in the AD brains was significantly higher than that in control brains. The grey matter/white matter uptake ratio was over ~2, the difference being significant for each 18F-radioligands [22]. A small 12-mer D-peptide (qshyrhispaqv), abbreviated as D1, could bind to Aβ in vitro in the sub-μmol/L range. 18F-labeling D1 derivative peptides was successfully obtained and showed a potential applicability for use in vivo by PET [23]. Besides, the tritium labelled D-peptide D3 (3H-D3) was prepared by SPPS and used to study its distribution and stability in different organisms (e.g., liver, kidney, plasma and gastrointestinal tract in mice), as well as its bioavailability with different administration routes [24]. The pharmacokinetic results showed D3 possessed a high stability and long biological half-life, representing a promising new compound class for drug development [25].
D3 can be chemically modulated to give its derivatives with improved properties. For example, a rationally designed derivative RD2, showed a long terminal half-life (more than 2 days) in plasma and high bioavailability with i.p. (intraperitoneal), s.c. (subcutaneous), or p.o. (per os, oral delivery) administration [8]. The further study of RD2 exhibited that RD2 could significantly increase the elimination efficiency of Aβ oligomers than D3. RD2 could improve cognitive performance of APP/PS1 mice and reduce insoluble Aβ levels in the brain without impacts on activity or anxiety [26]. One of the identified D-peptides, DB3, was found to eliminate Aβ oligomers, reduce Aβ-induced cytotoxicity and disassemble Aβ aggregates. In addition, the arginine-rich D3 can rapidly enter the brain by a direct penetration of the blood–brain barrier (BBB) to interact with amyloid plaques [27]. Besides, the optimization potential of linear D3 with free C-terminus (D3COOH) was performed by chemical modifications. The results showed that the charge increase and cyclization of D3 might improve the performance [28]. These excellent results indicated D3 and its derivatives were very promising drug candidates for Alzheimer's disease (Fig. 4). Remarkably, an Alzheimer's drug R2 (alias Contraloid or PRI-002) has already passed the phase I clinical trial (NCT03944460).
D3 and its derivatives were further modified into head-to-tail tandem D-peptides to improve the pharmacokinetic profiles. Both D3D3 and RD2D3 showed proteolytic stability in mouse plasma and organ homogenates, and even in murine and human liver microsomes. Both peptides were taken up into the brain by i.v. or i.p. administration [29]. The affinity of D3D3 towards Aβ could increase in comparison to D3 [20]. The head-to-tail tandem of DB3, DB3DB3, showed highly enhanced efficacy than DB3 [30]. Note that the cognitive deficit of Swedish and London mutations (APPSL) mice could be completely reversed by RD2 and D3D3 treatment [31].
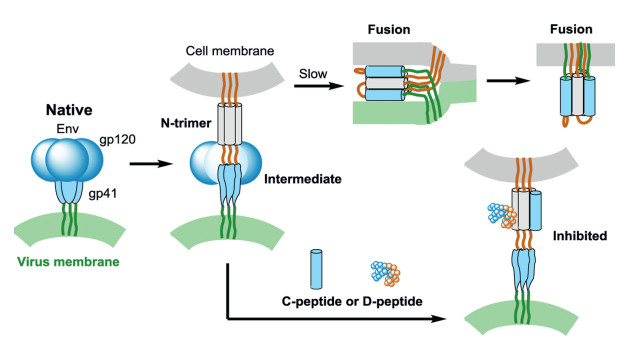
The chemical synthesis assisted mirror-image phage display technology was used to develop D-peptide binders to target membrane proteins. For example, HIV-1 envelope glycoprotein gp41 mediates the fusion of viral and cellular membranes and plays an important role in HIV entry (Fig. 5). Targeting the key HIVrelated membrane protein can inhibit HIV entry. The gp41 was considered as a potential drug target to block viral fusion/entry. To get a D-peptide inhibitor, the D-enantiomeric form of trimeric IQN17 (D-IQN17) was chemically synthesized and applied for the screening of L-peptide sequences that bind to D-IQN17 by a phage display library. Finally, D-peptides, the mirror-images of the phageexpressed peptides, were generated and identified with anti-HIV-1 activity by a HIV-1 infection assay. These short D-peptides can specifically bind to the gp41 hydrophobic pocket as shown by X-ray crystallography [32, 33].
The first generation D-peptides could only inhibit HIV-entry with a potency of IC50 at μmol/L levels. Therefore, Welch et al. combined the mirror-image phage display and structure-assisted design to improve the antiviral potency of D-peptides. One of Dpeptides pocket-specific inhibitors, shorten for PIE7, was obtained with up to 40, 000-fold potent inhibitory activity over the first generation D-peptides [34].
D-Peptide PIE7 showed moderate activity in the clinical neutralization test. Therefore, the third generation D-peptide PIE12, which was trimeric version of PIE7, was discovered with broadened inhibition potential and resistance to viral variants [35]. These D-peptide HIV inhibitors have shown specific and high affinity with HIV-1 gp41 and are considered as promising therapeutic anti-HIV agents [36, 37]. The D-peptide HIV entry inhibitor CPT31, an advanced version of PIE12, has initiated the phase I study. The development of D-peptide HIV-1 entry inhibitors showed again the efficiency of the mirror-image phage display method.
Recently, extracellular cysteine-rich domains (CRD) of fibroblast growth factor-inducible14 (Fn14), another membrane receptors, was chosen as the D-protein target. The D-Fn14 canonical CRD was prepared by SPPS and then mirror-image phage display strategy was used to afford a D-peptide ligand of Fn14, which enabled to target pancreatic and TNBC cancer cells [38]. In addition, the mirror-image mRNA display was also used to provide D-peptides to selectively target D-type CRD of Fn14 with powerful inhibition of the proliferation of activated HSCs [39]. These obtained D-peptide ligands showed strong binding affinity to Fn14 and high proteolytic resistance.
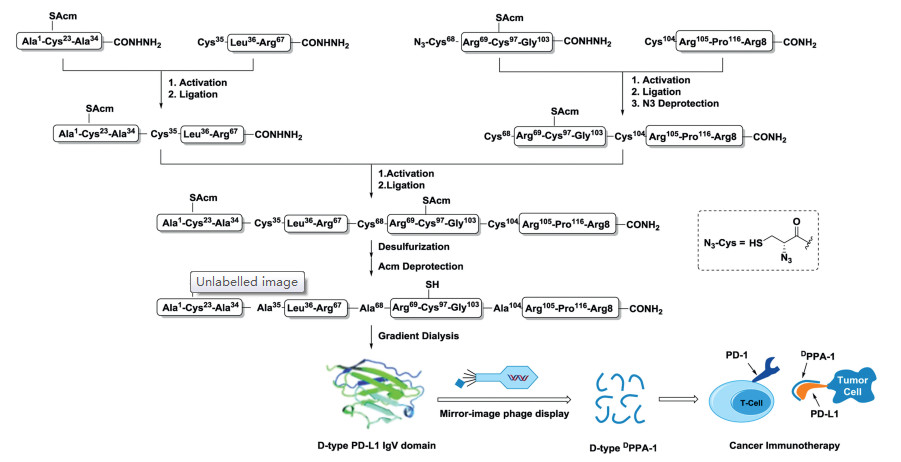
One key feature of mirror-image phage display is the use of the enantiomeric D-protein target as the key component for mirror-image phage display. These D-protein targets can only be obtained by chemical synthesis. Usually, SPPS chemistry can only provide peptides or small protein targets with about 50~60-residue [40, 41], such as the above mentioned SH3 domain, HIV-1 gp41 peptide, and Aβ. To prepare larger D-protein targets, chemical ligation techniques, such as native chemical ligation (NCL) [42-44] serine/threonine ligation (STL) [45], α-keto acid-hydroxylamine (KAHA) ligation [46] and diselenide-selenoester ligation (DSL) [47], have been developed to conjugate two or more short peptide segments into a longer peptide (Fig. 6). These powerful ligation methods have been widely used to generate D-proteins and greatly expanded the applications of mirror-image phage display in peptide drug discovery.

The tumor suppressor p53 transcriptionally regulates the gene expression to mediate DNA repair, cell growth, development, or apoptosis. MDM2 down-regulates the p53 activity by inhibiting p53 transcriptional activity and promoting p53 degradation. Restoring the p53 activity can inhibit the growth of tumors. Therefore, MDM2 has been considered as an important drug target for the development of anticancer antagonists. Initially, a potent L-peptide PMI (TSFAEYWNLLSP) was developed as an inhibitor of p53/MDM2 interaction by phage display method. PMI can bind to MDM2 with a high affinity (at nmol/L level), two orders of magnitude higher than that of the wild-type p53 peptide (ETFSDLWKLLPE). Unfortunately, PMI was susceptible to proteolysis, which dramatically reduced its therapeutic value [48].
To solve the in vivo proteolytic susceptibility of peptides, the Lu group used the chemical protein synthesis assisted mirror-image phage display strategy to give D-peptide ligands with high proteolytic resistance. First, the biotinylated D-MDM2 (25-109) was synthesized by native chemical ligation [49]. Then, a 12-mer phage display peptide library was constructed and then the mirror-image phage display was carried out to discover D-peptide antagonists of MDM2. Three D-peptide inhibitors, DPMI-α (tnwyanlekll), DPMI-β (tawyanfekllr) and DPMI-γ (dwwplafeallr), were successfully obtained with affinities of 219, 35 and 53 nmol/L, respectively. In addition, the Lu group designed a super-active Dpeptide DPMI-δ (6-F-Trp3/p-CF3-Phe7-DPMI-β) with an ultrahigh affinity of 220 pmol/L. X-ray crystal structure of MDM2/DPMI-δ complex indicated that DPMI-δ was a potent inhibitor of the p53- MDM2 interaction [50] (Fig. 7). Such D-peptides were fully resistant to proteolytic degradation and promising to be lead drug candidates for anticancer therapeutic research.
Note that the cell penetration of these DPMI peptides was poor, which hampered the therapeutic applicability of DPMI antagonists. To overcome the problem, peptide modification by combination of palmitoylated DPMI inhibitor of p53/MDM2 and RGD modified liposomes were performed to enhance tumor-targeting specificity of targeted molecular therapy. N-terminally palmitolated DPMI-β and the resultant c(RGDyK) decorated liposomes (RGD-liposomal pDP) showed almost 100% encapsulation efficiency and 10% loading efficiency [51].
The combination of chemical ligation and mirror-image phage display was extended the membrane receptor targets to provide D-peptide binders, such as the epidermal growth factor (EGF) and vascular endothelial growth factor A (VEGF-A). To find proteaseresistant D-peptides to disrupt the EGF-EGFR interaction, the Giralt group chemically synthesized the mirror-image protein D-EGF and then used mirror-image phage-display to select the D-peptides bound to natural EGF. The identified D-PI_4 can be efficiently disrupt the EGF–EGFR interaction [52].
To obtain D-peptide ligands targeting VEGF-A, the mirror-image form of VEGF-A was generated by sequential native chemical ligation (Fig. 8). Then, a 56-residue GB1 protein (B1 domain of streptococcal protein G) based peptide library was displayed on M13 phage. The GB1-based library was screened against the synthetic D-VEGF-A to give a peptide RFX001 with high binding affinity to D-VEGF-A. Finally, mirror-image RFX001 (D-RFX001) was prepared and identified to specifically bound with native L-VEGF-A with a high affinity (~ 85 nmol/L), but not D-VEGF-A. DRFX001 could inhibit the interaction of VEGF with VEGF receptor Flt1 (VEGFR1) [53]. Furthermore, by using the racemic crystallization strategy [54, 55], X-ray crystal structures of the heterochiral complex between D-RFX001 and L-VEGF-A were measured. The result identified that the D-antagonist bound to the region of VEGFA where VEGF-A interacts with its receptor VEGFR1 [53].

However, D-RFX001 is unstable at physiological temperatures (Tm = 33 ℃). To improve the thermal stability, D-RFX001 was then conveniently modified with an N-terminal Arg5 tag and extra five amino acids to give a stable D-RFX037 (Tm > 95 ℃). D-RFX037 was high affinity for VEGF-A (~6 nmol/L) and stable in mouse, monkey, and human plasma. Besides, D-RFX037 was nonimmunogenic in mice, whilst the L-RFX037 stimulated a strong immune response. These results confirm the potential utility of synthetic D-peptides as alternatives to therapeutic antibodies. These D-antagonists may have the potential as a new class of angiogenesis blockers for anticancer therapy [56].
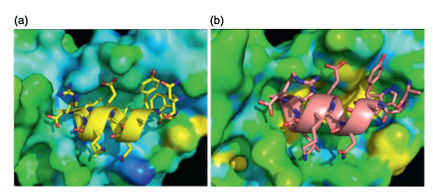
The combination of chemical protein synthesis and mirror-image phage display techniques was also employed to discover the hydrolysis resistant D-peptide antagonists targeting the PD-1/PDL1 pathway for cancer immunotherapy. The programmed-death ligand 1 (PD-L1) is one of important membrane protein target for checkpoint blockade therapy. The blockade of PD-L1 by extra antagonists can inhibit the immune-suppressing pathway to enhance anti-cancer immune responses of T cells for killing cancer cells, which has exhibited marked clinical benefit [57, 58]. Lots of antibody blockers have been developed to activate antitumor immunity. However, antibody drugs have some drawbacks in regard with cost and immunogenicity. Besides, antibodies can only block the function of PD-L1 on the tumor cell surface. The intracellular PD-L1, which can be active and redistribution to the cell surface to minimize the therapeutic benefits, is out of reach of antibody. Therefore, it is of importance to develop the new inhibitors against PD-L1 [59, 60]. Combining the native chemical ligation and mirror-image phage display, the first hydrolysis resistant D-peptide antagonist (DPPA-1, nyskptdrqyhf) was developed to target PD-L1 at an affinity of 0.51 μmol/L in vitro (Fig. 9). Tumor growth inhibition experiments of tumor-bearing mice indicated that the D-peptide DPPA-1 could effectively inhibit tumor growth and prolong animal survival by blocking the PD-1/PD-L1 interaction [61].
Besides, the recent advances in chemical protein synthesis, such as peptide hydrazide ligation [62-67], provided many mirror-image proteins that can be used as phage display targets for D-peptide inhibitors. For instance, the Kay group utilized native chemical ligation-desulfurization strategy to chemically synthesize the mirror-image inflammatory cytokine tumor necrosis factor alpha (TNFα), which plays a crucial role in the pathogenesis of chronic inflammatory disease [68]. Besides, the mirror-image ubiquitin chains [69-71], the mirror-image anaphylatoxin C5a [72], the mirror-image oncogenic KRas (G12 V) [73], the mirror-image Ig2 domain of Axl, one of the pivotal extracellular regions of the receptor involved in ligand binding [74], and even a D-membrane protein, the mirror-image M2 proton channel of influenza A virus [75] were also successfully obtained. The Oishi group prepared the TMR- and biotin-labeled XIAP BIR3 domains and their mirror-image proteins [76]. These work showed chemical ligation methods dramatically facilitate the preparation of D-proteins for mirror-image phage display.
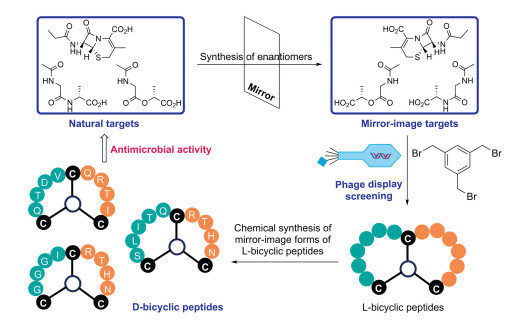
To increase the structural diversity of D-peptides, the more constrained bicyclic peptide libraries were employed during mirror-image phage display. In general, the bicyclic L-peptide library was first displayed as linear peptides containing three free Cys residues on the pIII coat protein of phage M13. The displayed peptide library was then chemically modified the reactive Cys with 1, 3, 5-tris(bromomethyl)benzene (TBMB) to give bicyclic peptide scaffolds. Bicyclic peptides have widely used as therapeutic drugs, research tools in biomedical and biological studies [77-79]. To obtain the bicyclic peptides composed of D-amino acids with antibacterial activity, the design and synthesis of enantiomers of target molecules (e.g., cephalosporin and N-acyl-D-Ala-D-Ala) was first carried out [80]. Then, phage display bicyclic peptide library was constructed, followed by affinity panning to obtain specific binders. After that, the D-forms of the selected L-peptides were synthesized by SPPS and identified by antimicrobial tests to provide the desired D-bicyclic peptides with antimicrobial activities against Staphylococcus aureus, Methicillin-resistant S. aureus, and vancomycin-resistant Enterococci, high stability, and no toxicity to mammalian cells (Fig. 10).
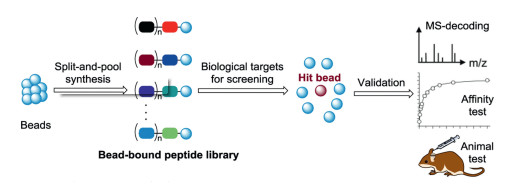
An alternative strategy for D-peptide ligands is the mirror-image OBOC method. Initially, OBOC method was mainly used to L-peptide ligands, where the OBOC peptide library composed of all L -amino acids was prepared by the split-and-pool method [81-84]. The solid-phase resin-bound peptides exist in the one-bead onecompound format, in which each bead carries multiple copies with a unique peptide sequence. The peptide library was screened for binding to a protein of interest. The hit peptides were determined by Edman sequencing or mass spectrometry (Fig. 11). When the D-amino acids were replaced to construct the library for screening, the OBOC method was referred as the mirror-image OBOC method (Fig. 12). In mirror-image OBOC, the native L-proteins, which can be prepared by the convenient biological expression technology, were used as screening targets. In addition, chemical protein synthesis enables the preparation of artificial L-protein targets (e.g. posttranslationally modified proteins). More importantly, the D-peptide libraries with any desired constructed structures can be easily synthesized by directly using D-amino acids [85-87]. The mirror-image OBOC approach can dramatically facilitate the discovery of D-peptide ligands.
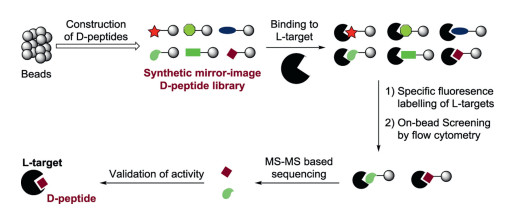
The mirror-image OBOC method is considered as a promising tool to generate D-peptide binders for drug development. As a case in point, the Pentelute group showed the de novo discovery of D-peptide binders for l-protein target [86]. In brief, the trypsin inhibitor EETI-II from Ecballium elaterium was chosen as a robust scaffold for the generation of a mirror-image OBOC 29- residue peptide library composed of D-amino acids. The D-peptide library containing beads were then incubated with fluorescent labelling protein targets (e.g. anti-hemagglutinin monoclonal antibody 12CA5 and thrombin). After that, a fluorescence-activated sorting (FACS) step was conducted by using flow cytometry to rapidly collect the beads that bound with the protein target. The sorted beads were treated with nucleophilic reagents to release the desired D-peptide ligands, which were verified by high-throughput de novo MS/MS peptide sequencing. Finally, the activity of D-type EETI-II binders was confirmed by orthogonal binding assays.
To reduce the false positives and biases from on-bead screening during the OBOC method, the Pentelute group developed a solution-phase affinity selection method that enables enrichment and sequencing of peptide binders. The screening platform relies on separation of bounded peptides by the high-performance sizeexclusion chromatography and high-through peptide sequencing by the tandem mass spectrometry to identify of high-affinity D-peptide ligands from focused libraries [88]. With these advances in the OBOC method, the OBOC-based direct screening of D-peptides would be a powerful method for the initial discovery of hit D-peptides [85, 87].
The computational method was developed to afford D-peptide inhibitors. For instance, using the available structure of a D-peptide inhibitor (D-RFX001) in complex with VEGF, the replacement by D-non-canonical amino acids was predicted in silico to improve binding affinity by as much -7.18 kcal/mol. These predictions were validated by experiments, approximately 105-fold improvement in Kd with VEGF [89]. Furthermore, to generate stable D-amino acid analogues of bioactive helical peptides, a method by using a mirror-image of the entire PDB was developed [90]. D-Peptide binders also have been computationally designed by the Lai group. They de novo designed novel D-peptide binders with α-helix structures, which can bound to TNFα. The D-peptides can inhibit the interaction of TNFα-TNFR1 with μmol/L affinity and reduce the activity of TNFα in vivo. The strategy would provide access to the discovery of D-peptide binders to inhibit protein-protein interactions [91].
D-Peptides have excellent performances in comparison with natural L-peptides, such as increased enzymatic stability and low immunogenicity, which make them attractive recognition molecules and therapeutic agents. However, the current therapeutic D-peptides are difficult to transport across cellular membranes. There are no endogenous transporters to deliver D-peptides. To achieve this, Zhou et al. reported a natural amino acid taurinemediated the cellular uptake of small D-peptides in mammalian cells [92]. In their study, a D-peptide conjugated with taurine can great increase the cellular uptake via both dynamin-dependent endocytosis and micropinocytosis from 118 μmol/L (without conjugation) to >1.6 μmol/L (after conjugation).
Bian et al. used aurous nanoparticles (AuNP) and a receptortargeted peptide to deliver D-peptides. They synthesized and functionalized the AuNP with a D-peptide p53 activator (DPA). The AuNP-based D-cargo can effectively enriched in tumor and internalized by cancer cells, reactivated p53 signaling to suppress tumor growth. Meanwhile, AuNP-DPA showed excellent biosafety without the common side effects. The AuNP-based deliver systems pave a way to deliver D-peptide PPI inhibitors into mammalian cells for cancer-targeted therapy (Fig. 13a) [93].

The Pentelute group employed protective antigen (PA) to deliver mirror-image peptide cargo (Fig. 13b). In the system, D-cargo was conjugated to the C-terminus of the N-terminal domain of lethal factor (LFN) which can binds stereo-specifically to PA pre-pore to transport various D-cargo into the cytosol of cells. The PA/LFN system can deliver different D-cargoes, such as mirror-image affibody and GB1 [94].
Schartmann et al. studied the blood-brain barrier penetration efficiencies of linear and cyclic D-peptides. A series of cyclic D-peptides were prepared by using cyclization strategy. The results showed that a head-to-tail cyclization of D3 can increase the peptide's efficiency to cross the murine BBB up to tenfold higher as compared to the corresponding linear peptides. Between them, cRD2D3 was found a suitable drug candidate with high proteolytic stability, good oral bioavailability, long lasting action and a favorable brain/plasma ratio [95].
D-Peptides are an exciting and attractive class of drug molecules that are protease-resistant, non-immunogenic, as well as possible orally bioavailable. Therefore, D-peptides are potential therapeutic and diagnosis agents for drug development. Many efforts have been made to develop practical and robust techniques for the highthroughput screening of D-peptide ligands and their delivery across the cell membranes.
Severalelegantmethods, such asmirror-imagephagedisplayand one-bead one-compound, have beenwidely used to affordD-peptide ligands. In combination with chemical synthesis of mirror-image proteins, these techniques have been applicable to relatively large protein targets. More importantly, many otherwise-difficult-toobtain protein targets, including site-specifically post-translationally modified proteins [71, 96-99] or membrane proteins [100-104], can be readily prepared thanks to the methodology advances in the chemical protein synthesis. The robust synthesis of protein targets and their corresponding D-enantiomers will greatly expand the applicationof the currenthigh-throughput screening strategies (e.g., mirror-imagephagedisplay, one-beadone-compound).Besides, the combination of mirror-image proteins with many recent developed high-throughput screening methods (for example, the random nonstandard peptide integrated discovery (RaPID) system for macrocyclic peptides), can be used to give D-peptide ligands with increased affinity [105].
It is alongtime dream to generate mirror-imagepe ptides/proteins via an artificial mirror-image synthetic biology system where there are mirror-image Escherichia coli, mirror-image bacteriophage, and others [106-109]. One solution to the construction of a functional mirror-image life system is the bottom-up approach [110] (Fig. 14). Specifically, a mirror-image phage will be artificially synthesized by using chemical protein synthesis toolbox and other advanced techniques. And then the screening was performed by using a normal "mirror-image phage" display to give D-peptides (Fig. 15).
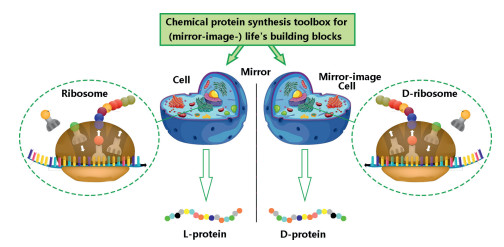

The critical challenge of mirror-image life is to establish full of chirally-flipped cell-like biological systems starting from molecular building blocks. Owing to the development of chemical protein synthesis, a series of significant preliminary explorations have been made to construct mirror-image molecular biology systems. For instance, the Kent's group achieved the total chemical synthesis of D-type HIV-1 protease with specificity of the D-peptide substrate [111]. The Kay group studied chaperoneassisted folding of chemically synthetic D-proteins [112]. The Liu group and Zhu group reported an outstanding work on chemical synthesis of mirror-image polymerase ASFV pol X. They achieved the mirror-image genetic replication/transcription in the central dogma [113]. Furthermore, the chemical synthetic routes for thermostable d-Dpo4 have been established and the mirror-image PCR was also successfully achieved [114-117]. Besides, the Zheng group prepared the mirror-image hydrophobic membrane protein M2 ion channel of influenza A virus [75]. These pioneering works pave a way to the mirror-image lives. The building of a fully mirror-image life system would open new avenues to create mirror-image peptides/proteins for vast applications in materials and pharmaceutical sciences.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This study was supported by the National Key R & D Program of China (No. 2019YFA0706902), National Natural Science Foundation of China (Nos. U1732161 and 91753120), and Science and Technological Fund of Anhui Province for Outstanding Youth (No. 1808085J04).
[1] T.T. Hansel, H. Kropshofer, T. Singer, J.A. Mitchell, A.J.T. George, Nat. Rev. Drug Discov. 9 (2010) 325-338. doi: 10.1038/nrd3003
M. Gongora-Benitez, J. Tulla-Puche, F. Albericio, Chem.Rev.114 (2014) 901-926. doi: 10.1021/cr400031z
A. Henninot, J.C. Collins, J.M. Nuss, J. Med. Chem. 61 (2018) 1382-1414. doi: 10.1021/acs.jmedchem.7b00318
(a) W. Liu, C. Wu, Chin. Chem. Lett. 29 (2018) 1063-1066;
(b) X. Li, Y. Zou, H.G. Hu, Chin. Chem. Lett. 29 (2018) 1088-1092;
(c) Y. Jiang, H. Long, Y. Zhu, Y. Zeng, Chin. Chem. Lett. 29 (2018) 1067-1073.
Q. Qu, S. Gao, F. Wu, et al., Angew. Chem. Int. Ed. 132 (2020) 6093-6101. doi: 10.1002/ange.201915358
Y. Li, K.A. Clark, Z. Tan, Chin. Chem. Lett. 29 (2018) 1074-1078. doi: 10.1016/j.cclet.2018.05.027
J.L. Lau, M.K. Dunn, Bioorg. Med. Chem. 26 (2018) 2700-2707. doi: 10.1016/j.bmc.2017.06.052
L.H.E. Leithold, N. Jiang, J. Post, et al., Pharm. Res. 33 (2016) 328-336. doi: 10.1007/s11095-015-1791-2
N. Sun, S.A. Funke, D. Willbold, Mini-Rev. Med. Chem. 12 (2012) 388-398. doi: 10.2174/138955712800493942
M. Cardó-Vila, R.J. Giordano, R.L. Sidman, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 5118-5123. doi: 10.1073/pnas.0915146107
X. Wang, Y. Qiao, I.A. Asangani, et al., Cancer Cell 31 (2017) 532-548. doi: 10.1016/j.ccell.2017.02.017
M. Chorev, M. Goodman, Acc. Chem. Res. 26 (1993) 266-273. doi: 10.1021/ar00029a007
C. Li, M. Pazgier, J. Li, et al., J. Biol. Chem. 285 (2010) 19572-19581. doi: 10.1074/jbc.M110.116814
J.K. Scott, G.P. Smith, Science 249 (1990) 386-390. doi: 10.1126/science.1696028
T.N.Schumacher, L.M. Mayr, D.L. Minor Jr, et al., Science 271 (1996) 1854-1857. doi: 10.1126/science.271.5257.1854
D.H. Cribbs, C.J. Pike, S.L. Weinstein, P. Velazquez, C.W. Cotman, J. Biol. Chem. 272 (1997) 7431-7436. doi: 10.1074/jbc.272.11.7431
(a) K. Wiesehan, K. Buder, R.P. Linke, et al., ChemBioChem 4 (2003) 748-753;
(b) C. Zuo, S. Tang, Y.Y. Si, et al., Org. Biomol. Chem. 14 (2016) 5012-5018.
K. Wiesehan, D. Willbold, ChemBioChem 4 (2003) 811-815. doi: 10.1002/cbic.200300570
S.A. Funke, D. Willbold, Mol. Biosyst. 5 (2009) 783-786. doi: 10.1039/b904138a
T. Dunkelmann, K. Teichmann, T. Ziehm, et al., Neuropeptides 67 (2018) 27-35. doi: 10.1016/j.npep.2017.11.011
S.A. Funke, T. van Groen, I. Kadish, et al., ACS Chem. Neurosci. 1 (2010) 639-648. doi: 10.1021/cn100057j
B. Gulyas, C. Spenger, Z. Beliczai, et al., Neurochem. Int. 60 (2012) 153-162. doi: 10.1016/j.neuint.2011.10.010
M. Jahan, S. Nag, R. Krasikova, et al., Nucl. Med. Biol. 39 (2012) 315-323. doi: 10.1016/j.nucmedbio.2011.09.008
N. Jiang, L.H.E. Leithold, J. Post, et al., PLoS One 10 (2015) e0128553. doi: 10.1371/journal.pone.0128553
A. Elfgen, B. Santiago-Schuebel, L. Gremer, J. Kutzsche, D. Willbold, Euro. J. Pharm. Sci. 107 (2017) 203-207. doi: 10.1016/j.ejps.2017.07.015
T. van Groen, S. Schemmert, O. Brener, et al., Sci. Rep. 7 (2017) 16275. doi: 10.1038/s41598-017-16565-1
N. Jiang, D. Frenzel, E. Schartmann, et al., Biochim. Biophys. Acta 1858 (2016) 2717-2724. doi: 10.1016/j.bbamem.2016.07.002
T. Ziehm, O. Brener, T. van Groen, et al., ACS Chem. Neurosci. 7 (2016) 1088-1096. doi: 10.1021/acschemneuro.6b00047
L.H.E. Leithold, N. Jiang, J. Post, et al., Euro. J. Pharm. Sci. 89 (2016) 31-38. doi: 10.1016/j.ejps.2016.04.016
A.N. Klein, T. Ziehm, M. Tusche, et al., PLoS One 11 (2016) e0153035. doi: 10.1371/journal.pone.0153035
J. Kutzsche, S. Schemmert, M. Tusche, et al., Molecules 22 (2017) 1693-1703. doi: 10.3390/molecules22101693
D.M. Eckert, V.N. Malashkevich, L.H. Hong, P.A. Carr, P.S. Kim, Cell 99 (1999) 103-115. doi: 10.1016/S0092-8674(00)80066-5
D.M. Eckert, V.N. Malashkevich, P.S. Kim, J. Mol. Biol. 284 (1998) 859-865. doi: 10.1006/jmbi.1998.2214
B.D. Welch, A.P. VanDemark, A. Heroux, C.P. Hill, M.S. Kay, Proc.Natl.Acad. Sci. U. S. A. 104 (2007) 16828-16833. doi: 10.1073/pnas.0708109104
B.D. Welch, J.N. Francis, J.S. Redman, et al., J. Virol. 84 (2010) 11235-11244. doi: 10.1128/JVI.01339-10
R.S. Lopes, M.A. Freitas Queiroz, S.T. Monteiro Gomes, et al., Biotechnol. Adv. 36 (2018) 1847-1854. doi: 10.1016/j.biotechadv.2018.07.003
S. Delhalle, J.C. Schmit, A. Chevigne, Int. J. Mol. Sci. 13 (2012) 4727-4794. doi: 10.3390/ijms13044727
Z. Li, J. Xie, S. Peng, et al., Bioconjugate Chem. 28 (2017) 2167-2179. doi: 10.1021/acs.bioconjchem.7b00326
L. Huang, J. Xie, Q. Bi, et al., Mol. Pharm. 14 (2017) 1742-1753. doi: 10.1021/acs.molpharmaceut.6b01174
J.S. Zheng, S. Tang, Y.C. Huang, L. Liu, Acc. Chem. Res. 46 (2013) 2475-2484. doi: 10.1021/ar400012w
S.B. Kent, Chem. Soc. Rev. 38 (2009) 338-351. doi: 10.1039/B700141J
V. Agouridas, O. El Mahdi, V. Diemer, et al., Chem. Rev.119 (2019) 7328-7443. doi: 10.1021/acs.chemrev.8b00712
P.E. Dawson, T.W. Muir, I. Clark-Lewis, S.B. Kent, Science 266 (1994) 776-779. doi: 10.1126/science.7973629
C. Zuo, B. Zhang, B. Yan, J.S. Zheng, Org. Biomol. Chem. 17 (2019) 727-744. doi: 10.1039/C8OB02610F
H. Liu, X. Li, Acc. Chem. Res. 51 (2018) 1643-1655. doi: 10.1021/acs.accounts.8b00151
J.W. Bode, Acc. Chem. Res. 50 (2017) 2104-2115. doi: 10.1021/acs.accounts.7b00277
S.S. Kulkarni, J. Sayers, B. Premdjee, R.J. Payne, Nat. Rev. Chem. 2 (2018) 0122. doi: 10.1038/s41570-018-0122
M. Pazgier, M. Liu, G. Zou, et al., Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 4665-4670. doi: 10.1073/pnas.0900947106
M. Liu, C. Li, M. Pazgier, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 14321-14326. doi: 10.1073/pnas.1008930107
C. Zhan, L. Zhao, X. Wei, et al., J. Med. Chem. 55 (2012) 6237-6241. doi: 10.1021/jm3005465
C. Li, J. Shen, X. Wei, C. Xie, W. Lu, J. Drug Target. 20 (2012) 264-271. doi: 10.3109/1061186X.2011.645162
C. Díaz-Perlas, M. Varese, S. Guardiola, et al., ChemBioChem 20 (2019) 2079-2084. doi: 10.1002/cbic.201900355
K. Mandal, M. Uppalapati, D. Ault-Riche, et al., Proc. Natl. Acad. Sci. U. S. A.109 (2012) 14779-14784. doi: 10.1073/pnas.1210483109
T.O. Yeates, S.B. Kent, Ann. Rev. Biophys. 41 (2012) 41-61. doi: 10.1146/annurev-biophys-050511-102333
R. Okamoto, K. Mandal, M.R. Sawaya, et al., Angew. Chem. Int. Ed. 53 (2014) 5194-5198.
M. Uppalapati, D.J. Lee, K. Mandal, et al., ACS Chem. Biol.11 (2016) 1058-1065. doi: 10.1021/acschembio.5b01006
G. Sonpavde, New Engl. J. Med. 376 (2017) 1073-1074. doi: 10.1056/NEJMe1701182
J.R. Brahmer, S.S. Tykodi, L.Q.M. Chow, et al., New Engl. J. Med. 366 (2012) 2455-2465. doi: 10.1056/NEJMoa1200694
H. Yao, J. Lan, C. Li, et al., Nat. Biomed. Eng. 3 (2019) 306-317. doi: 10.1038/s41551-019-0375-6
J. Zhang, X. Bu, H. Wang, et al., Nature 553 (2018) 91-95. doi: 10.1038/nature25015
H.N. Chang, B.Y. Liu, Y.K. Qi, et al., Angew. Chem. Int. Ed. 54 (2015) 11760-11764. doi: 10.1002/anie.201506225
G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. doi: 10.1002/anie.201100996
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. doi: 10.1002/anie.201203843
J.S. Zheng, S. Tang, Y.K. Qi, Z.P. Wang, L. Liu, Nat. Protoc. 8 (2013) 2483-2495. doi: 10.1038/nprot.2013.152
S. Tang, Y.Y. Si, Z.P. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 5713-5717. doi: 10.1002/anie.201500051
L. Liu, Isr. J. Chem. 59 (2019) 64-70. doi: 10.1002/ijch.201800135
(a) J. Liu, S. Dong, Chin. Chem. Lett. 29 (2018) 1131-1134;
(b) S. Xu, Z. Zhao, J. Zhao, Chin. Chem. Lett. 29 (2018) 1009-1016;
(c) J. Yang, J. Zhao, Sci. China Chem. 61 (2018) 97-112;
(d) C.C. Chen, S. Gao, H.S. Ai, et al., Sci. China Chem. 61 (2018) 702-707.
M.E. Petersen, M.T. Jacobsen, M.S. Kay, Org. Biomol. Chem. 14 (2016) 5298-5303. doi: 10.1039/C6OB00824K
M. Pan, S. Gao, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 7429-7435. doi: 10.1021/jacs.6b04031
S. Gao, M. Pan, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 14497-14502. doi: 10.1021/jacs.6b09545
S. Tang, L.J. Liang, Y.Y. Si, et al., Angew. Chem. Int. Ed. 56 (2017) 13333-13337. doi: 10.1002/anie.201708067
C. Zuo, W.W. Shi, X.X. Chen, et al., Sci. China Chem. 62 (2019) 1371-1378. doi: 10.1007/s11426-019-9513-2
A.M. Levinson, J.H. McGee, A.G. Roberts, et al., J. Am. Chem. Soc. 139 (2017) 7632-7639. doi: 10.1021/jacs.7b02988
L. De Rosa, R. Di Stasi, L.D. D'Andrea, Tetrahedron 75 (2019) 894-905. doi: 10.1016/j.tet.2019.01.005
Q.Y. Guo, L.H. Zhang, C. Zuo, et al., Protein Cell 10 (2019) 211-216. doi: 10.1007/s13238-018-0536-5
K. Shu, N. Iwamoto, K. Honda, et al., Bioconjugate Chem. 30 (2019) 1395-1404. doi: 10.1021/acs.bioconjchem.9b00154
C. Heinis, T. Rutherford, S. Freund, G. Winter, Nat. Chem. Biol. 5 (2009) 502-507. doi: 10.1038/nchembio.184
I. Rentero Rebollo, C. Heinis, Methods 60 (2013) 46-54. doi: 10.1016/j.ymeth.2012.12.008
K. Deyle, X.D. Kong, C. Heinis, Acc. Chem. Res. 50 (2017) 1866-1874. doi: 10.1021/acs.accounts.7b00184
E. Adaligil, K. Patil, M. Rodenstein, K. Kumar, ACS Chem. Biol. 14 (2019) 1498-1506. doi: 10.1021/acschembio.9b00234
K.S. Lam, S.E. Salmon, E.M. Hersh, et al., Nature 354 (1991) 82-84. doi: 10.1038/354082a0
S.W. Millward, R.K. Henning, G.A. Kwong, et al., J. Am. Chem. Soc. 133 (2011) 18280-18288. doi: 10.1021/ja2064389
J.E. Jee, J. Lim, Y.S. Ong, et al., Org. Biomol. Chem. 14 (2016) 6833-6839. doi: 10.1039/C6OB01104G
Y.S. Ong, L. Gao, K.A. Kalesh, et al., Curr. Top. Med. Chem.17 (2017) 2302-2318.
V.V. Komnatnyy, T.E. Nielsen, K. Qvortrup, Chem. Commun. 54 (2018) 6759-6771. doi: 10.1039/C8CC02486C
Z.P. Gates, A.A. Vinogradov, A.J. Quartararo, et al., Proc. Natl. Acad. Sci. U. S. A. 115 (2018) E5298-E5306. doi: 10.1073/pnas.1722633115
D. Pei, G.A. Kubi, Expert. Opin. Drug Dis. 14 (2019) 1097-1102. doi: 10.1080/17460441.2019.1647164
F. Touti, Z.P. Gates, A. Bandyopdhyay, G. Lautrette, B.L. Pentelute, Nat. Chem. Biol. 15 (2019) 410-418. doi: 10.1038/s41589-019-0245-2
M. Garton, M. Sayadi, P.M. Kim, PLoS One 12 (2017) e0187524. doi: 10.1371/journal.pone.0187524
M. Garton, S. Nim, T.A. Stone, et al., Proc. Natl. Acad. Sci. U. S. A. 115 (2018) 1505-1510. doi: 10.1073/pnas.1711837115
W. Yang, Q. Zhang, C. Zhang, et al., FEBS Lett. 593 (2019) 1292-1302. doi: 10.1002/1873-3468.13444
J. Zhou, X. Du, J. Li, N. Yamagata, B. Xu, J. Am. Chem. Soc. 137 (2015) 10040-10043. doi: 10.1021/jacs.5b06181
Z. Bian, J. Yan, S. Wang, et al., Theranostics 8 (2018) 5320-5335. doi: 10.7150/thno.27165
A.E. Rabideau, X. Liao, B.L. Pentelute, Chem. Sci. 6 (2015) 648-653. doi: 10.1039/C4SC02078B
E. Schartmann, S. Schemmert, T. Ziehm, et al., Eur. J. Pharm. Sci. 114 (2018) 93-102. doi: 10.1016/j.ejps.2017.12.005
P. Gopinath, S. Ohayon, M. Nawatha, A. Brik, Chem. Soc. Rev. 45 (2016) 4171-4198. doi: 10.1039/C6CS00083E
Y.K. Qi, Y.Y. Si, S.S. Du, et al., Sci. China Chem. 62 (2019) 299-312. doi: 10.1007/s11426-018-9401-8
M. Pan, Q. Zheng, S. Gao, et al., CCS Chem. 1 (2019) 476-489. doi: 10.31635/ccschem.019.20190001
L.J. Liang, Y. Si, S. Tang, et al., Chin. Chem. Lett. 29 (2018) 1155-1159. doi: 10.1016/j.cclet.2018.03.022
J.S. Zheng, M. Yu, Y.K. Qi, et al., J. Am. Chem. Soc. 136 (2014) 3695-3704. doi: 10.1021/ja500222u
C. Zuo, S. Tang, J.S. Zheng, J. Pept. Sci. 21 (2015) 540-549. doi: 10.1002/psc.2721
J.S. Zheng, Y. He, C. Zuo, et al., J. Am. Chem. Soc. 138 (2016) 3553-3561. doi: 10.1021/jacs.6b00515
J.B. Li, S. Tang, J.S. Zheng, C.L. Tian, L. Liu, Acc. Chem. Res. 50 (2017) 1143-1153. doi: 10.1021/acs.accounts.7b00001
D.L. Huang, C. Montigny, Y. Zheng, et al., Angew. Chem. Int. Ed. 59 (2020) 5178-5184. doi: 10.1002/anie.201914836
Y. Huang, M.M. Wiedmann, H. Suga, Chem. Rev. 119 (2018) 10360-10391.
J. Bohannon, Wired (2010). http://www.wired.com/2010/11/ff_mirrorlife/.
Z.C. Li, B.C. Zhang, C. Zuo, L. Liu, Chin. J. Org. Chem. 38 (2018) 2412-2419. doi: 10.6023/cjoc201804014
R. Meledin, A. Brik, Cell Chem. Biol. 26 (2019) 616-619. doi: 10.1016/j.chembiol.2019.04.013
S.B.H. Kent, Protein Sci. 28 (2019) 313-328. doi: 10.1002/pro.3533
H. Mutschler, T. Robinson, T.Y.D. Tang, S. Wegner, ChemBioChem 20 (2019) 2533-2534. doi: 10.1002/cbic.201900507
R.C. Milton, S.C. Milton, S.B. Kent, Science 256 (1992) 1445-1448. doi: 10.1126/science.1604320
M.T. Weinstock, M.T. Jacobsen, M.S. Kay, Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 11679-11684. doi: 10.1073/pnas.1410900111
Z. Wang, W. Xu, L. Liu, T.F. Zhu, Nat. Chem. 8 (2016) 698-704. doi: 10.1038/nchem.2517
W. Jiang, B. Zhang, C. Fan, et al., Cell Discov. 3 (2017) 17037.
B. Zhang, Q. Deng, C. Zuo, et al., Angew. Chem. Int. Ed 58 (2019) 12231-12237. doi: 10.1002/anie.201905149
J. Weidmann, M. Schnoelzer, P.E. Dawson, J.D. Hoheisel, Cell Chem. Biol. 26 (2019) 645-651. doi: 10.1016/j.chembiol.2019.02.008
A. Pech, J. Achenbach, M. Jahnz, et al., Nucleic Acids Res. 45 (2017) 3997-4005. doi: 10.1093/nar/gkx079

Figure 3 The principle of mirror-image phage display. D-Protein targets for phage-display screening were chemically synthesized. L-Peptides that specifically bind to the D-target can be obtained by phage display library biopanning. The mirror-image forms (D-peptides) of the selected L-peptides were prepared by SPPS, which would specifically bind to the L-target. The biopanning process (repeat 3-4 times) usually needs five steps to finish a cycle.

Figure 4 (a) The structure of Aβ-D3 complex; (b) Peptide sequences of D3 and its derivatives; (c) Chemical structure of D3.

Figure 5 HIV entry and inhibitory. HIV Env contains gp120 (green) and gp41 (blue). CD4 and a co-receptor activate gp41, mediate the HIV fusion and induce formation of prehairpin intermediate. The N-terminal of gp41 forms a trimeric coiled coil (N-trimer, gray). C-peptide or D-peptide inhibitors can bind to the N-trimer to prevent trimer-ofhairpins formation and membrane fusion.

Figure 7 (a) The interface of MDM2-DPMI-α peptide complex (PDB: 3LNJ) and (b) MDM2-DPMI-δ peptide complex (PDB: 3TPX). The D-peptides (cartoons) and its side chains (sticks).

Figure 8 Chemical synthesis of homodimeric mirror-image VEGF-A by native chemical ligation and D-antagonists for VEGF-A.

Figure 9 Mirror-image phage display for the hydrolysis resistant D-peptide antagonists to target immune checkpoint protein PD-L1.

Figure 13 D-Peptide delivery. (a) The mechanism of the enhanced tumor targeting of AuNP-DPA by RGD conjugation and stimuli-responsive release of its D-cargo. (b) Delivery of LFN-conjugated D-cargo by protective antigen (PA).

Figure 14 The chemical protein synthesis assisted construction D-ribosome based protein factory for D-peptides/proteins.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:





