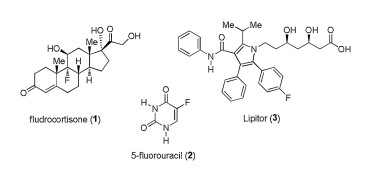
Figure 1.
Structures of fluorine-containing drugs fludrocortisone (1), 5-fluorouracil (2) and Lipitor (3).
Fluorine-containing drugs approved by the FDA in 2019
Haibo Mei , Attila Márió Remete , Yupiao Zou , Hiroki Moriwaki , Santos Fustero , Lorand Kiss , Vadim A. Soloshonok , Jianlin Han

Since the groundbreaking discovery and introduction to the market of fludrocortisone 1 (Fig. 1) [1] and 5-fluorouracil 2 [2], in the mid-1950th, biological applications of fluorine have become a mature field of mainstream multidisciplinary research advancing healthcare industry. In fact, fluorine-containing molecules constitute more than 50% of so-called blockbuster drugs [3] with Lipitor (3) [4] being, perhaps, the most profitable medicine ever introduced to the market. Presently, fluorine-scan/fluorine editing is virtually a routine step in the drug-discovery process and more generally, in bioorganic and medicinal chemistry explorations [5]. Various aspects of fluorine applications in the healthcare industry and drug design have already been covered by several reviews [6]. However, the area of fluorine-containing pharmaceuticals is currently undergoing very rapid development deserving timely updates awaited for by the multidisciplinary scientific community. To address this issue, last year we have initiated the mini-review series covering the most recent developments on the market of fluorinated drugs. The first review, under the title: "FluorineContaining Drugs Approved by the FDA in 2018" [7], was well received by the readers, generating keen interest, as can be inferred from the number of downloads and citations. In the present work, we profile new 11 fluorine-containing drugs approved for commercial use by the FDA in 2019. These include lumateperone (for the treatment of schizophrenia), ubrogepant (migraine), siponimod (relapsing form of multiple sclerosis), lemborexant (insomnia), upadacitinib (rheumatoid arthritis), pretomanid (antituberculosis), alpelisib (breast cancer), selinexor (cancer), entrectinib (lymphoma kinase inhibitor), pexidartinib (cancer) and lasmiditan (serotonin receptor antagonist). For each compound we discuss the discovery, biological properties and the synthetic route used for production, emphasizing the source of fluorinecontaining moieties.
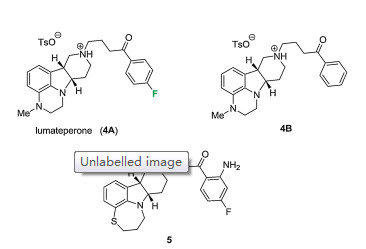
Lumateperone (ITI-007, 4A) was developed by Intra-Cellular Therapies, licensed from Bristol-Myers Squibb. Its FDA approval for the treatment of schizophrenia in adults was granted in December 2019 [8]. Lumateperone is currently tested for its applicability in bipolar disorder.
The structure of lumateperone was first covered in a patent filed by DuPont Pharmaceuticals in 2000 (Fig. 2) [9]. Later, researchers of Bristol-Myers Squibb published the synthesis of selective 5- HT2a/D2 receptor antagonists, including cis-(8a, 12a)-6, 7, 8a, 9, 10, 11, 12, 12a-octahydro-5H-pyrido[4, 3-b][1, 4]thiazepino[2, 3, 4-hi]indole 5 (Fig. 2) [10]. Systematic variation of the heterocyclic cores (ring sizes, heteroatoms, and stereochemistry), as well as the side chains (ring systems and linker types), lead to 2, 3, 6b, 9, 10, 10ahexahydro-1H, 7H-pyrido[30, 40:4, 5]pyrrolo[1, 2, 3-de]quinoxalines such as lumateperone (4A) with improved physicochemical and pharmacological properties [11]. The fluorine substitution is very important. Compound 4A is a potent antagonist of serotonin receptor 5-HT2a (Ki = 0.54 nmol/L), while more than ten folds of Ki value (5.8) is observed for the non-fluorinated compound 4B [11].
At dopamine-D2 receptors, lumateperone acts as a postsynaptic antagonist and pre-synaptic partial agonist (Ki = 32 nmol/L for D2 receptors and Ki = 52 nmol/L for D1 receptors) with mesolimbic/mesocortical selectivity. Its affinity to serotonin transporters (SERTs) is Ki = 62 nmol/L. The affinity differences enable the treatment of different medical conditions in a dosedependent manner [12]. Low doses selectively block 5-HT2a receptors, which was expected to reduce behavioral disturbances in dementia and insomnia in neuropsychiatric disorders [12]. Unfortunately, Phase III study of the treatment of agitation in patients with presumable Alzheimer's disease was discontinued because it was not likely to meet its primary endpoint. Although the Phase II study of patients with primary insomnia, as expected, showed significant improvement [12, 13], no further steps were taken.
In higher doses, 5-HT2a receptors are fully saturated, while dopamine-D2 receptor and SERT interactions can be titrated to desired levels. Since biochemical markers of presynaptic D2 activity (e.g., dopamine turnover) are unchanged, extra pyramidal side effects can be avoided. Block of dopamine activity in the pituitary gland, which causes prolactin increase, is avoided thanks to the mesolimbic/mesocortical selectivity on dopamine receptors. Clinical trials using higher lumateperone doses focused on schizophrenia and bipolar disorder. In both cases, lumateperone was well-tolerated (its safety profile is similar to that of placebo) and demonstrated statistically significant advantages over risperidone (a common antipsychotic) on key safety and tolerability parameters including glucose, lipids and weight gain. In contrast with common antipsychotics, lumateperone also improved the negative symptoms of schizophrenia [12, 14, 15]. In the case of schizophrenia, FDA approved lumateperone for the treatment of adults. In the case of bipolar disorder, two Phase III studies of lumateperone monotherapy had controversial results: one had positive top-line results, while the other study failed (partly because of high placebo response). A third Phase III study, which uses lumateperone as an adjunctive therapy to lithium or valproate, is still in progress [16].
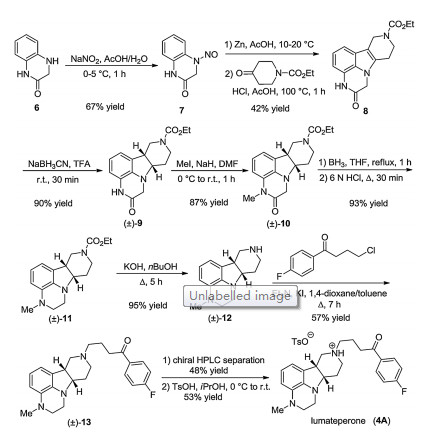
The synthesis of lumateperone started from 3, 4-dihydroquinoxalin-2(1H)-one. The first step is N-nitrosation yielding nitrosoquinoxalinone derivative 7 (Scheme 1). Compound 7 was reduced with Zn in AcOH to a hydrazine derivative, which was subjected to Fischer indole synthesis with ethyl 4-oxopiperidine- 1-carboxylate without isolation, resulting in tetracyclic derivative 8. Its olefin bond was reduced cis-selectively with NaBH3CN in TFA furnishing indoline derivative (±)-9. Amide methylation, boranemediated reduction of the amide moiety, and subsequent Ndeprotection in the presence of KOH in nBuOH yielded compound (±)-12. The 4-fluorobutyrophenone side chain was introduced by N-alkylation. The obtained racemic compound (±)-13 was resolved with chiral HPLC and the resulting (6bR, 10aS)-13 was transformed into lumateperone (4A) with TsOH in isopropyl alcohol [11].
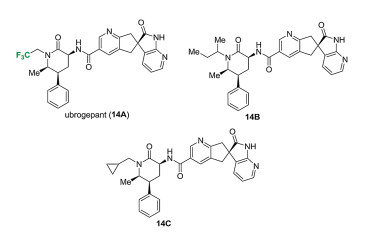
Ubrogepant (MK-1602, 14A) was developed by Allergan, licensed from Merck. It was approved by FDA in December 2019 for the acute (immediate) treatment of migraine (Fig. 3). Merck also prepared several non-fluorinated compounds (14B and 14C) as the piperidinone carboxamide azaindane calcitonin gene-related peptide (CGRP) receptor antagonists [17].
Available data allow to conclude that migraine is a disorder of the nervous system. Calcitonin gene-related peptide seems to be a key neuropeptide involved. For example, circulating levels of CGRP increase during spontaneous migraine attacks. In addition, exogenous CGRP can trigger acute headache and delayed migraine-like attacks in migraineurs. Taking these into account, suppression of excessive CGRP signaling seemed to be a promising new opportunity to treat migraine. Various small-molecule CGRP receptor antagonists ('gepants'), such as olcegepant, telcagepant, MK-3207, ubrogepant, and rimegepant were synthesized [18].
Importantly, the majority of clinical trials of gepants proved positive. However, development of olcegepant was discontinued, because it required intravenous administration (poor oral bioavailability). Development of telcagepant and MK-3207 was suspended, too, because of hepatotoxicity concerns [17, 18]. In contrast, clinical trials of the chemically distinct ubrogepant showed good efficacy, safety, and tolerability (no hepatotoxicity side effects) [19, 20].
The structure and preparation of ubrogepant and related molecules were first covered in a patent filed by Merck in 2012 [21]. The two halves of ubrogepant (a spirocyclic carboxylic acid and an aminopiperidinone molecule) were prepared separately, then a coupling agent was used to create the amide bond between them. Two additional Merck patents [22, 23] contain slight variations of the above synthesis. A full description of these synthetic improvements including yields was published in 2017 [24]. In this improved synthesis, aminopiperidinone salt 22 is prepared in the following steps. N-Protected serine ester (±)-15 is subjected to a dehydration/Michael addition sequence to obtain compound 17. This was initially accomplished with 4-bromophenylacetone, necessitating a debromination step, whereas phenylacetone (a schedule II controlled substance by US DEA) was used later for larger scales without debromination. Compound 17 was cyclized into product 18, a mixture of C-3 epimers, by dynamic kinetic transamination using an enzyme optimized for this sole reaction. This C-3 position epimerizes under basic conditions, favoring the desired configuration, that is N-alkylation was performed on the mixture of 18. This alkylation step required the highly reactive reagent CF3CH2OTf, and careful optimization was necessary to minimize alkylation on both N-atoms. The obtained crude product 19 was subjected to selective Boc deprotection. First, unreacted 18 was selectively deprotected and removed, then compound 19 was deprotected and separated from double-alkylated byproduct 20. The obtained amine mixture (21) was subjected to crystallization inducing a diastereoselective transformation (p-toluic acid to form a crystalline salt and 3, 5- dichlorosalicylaldehyde catalyst for C-3 epimerization via reversible imine formation) (Scheme 2).
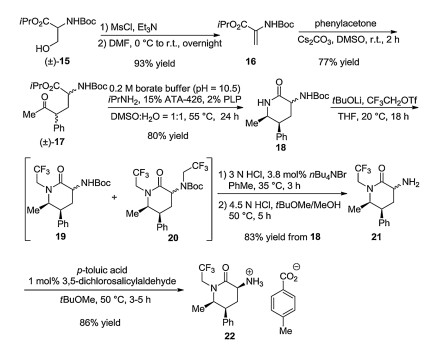
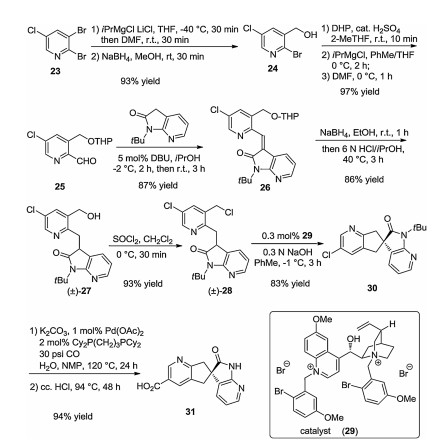
Preparation of spirocyclic carboxylic acid 31 started from 2, 3- dibromo-5-chloropyridine 23 (Scheme 3). After selective halogen– lithium exchange, the formyl group was introduced at C-3 followed by its immediate reduction with NaBH4. The resulting compound 24 underwent O-protection by 3, 4-dihydro-2H-pyran (DHP), then a second selective halogen–lithium exchange led to the introduction of the formyl group at C-2. Condensation of aldehyde 25 with N-(t-butyl)-azaindolone provided Z-alkene 26. Reduction of the newly created C=C double bond followed by O-deprotection yielded racemic alcohol (±)-27. The formed OH group was replaced with Cl upon reaction with SOCl2 affording compound (±)-28. Enantioselective spirocyclization of the latter achieved in the presence of chiral phase transfer catalyst 29 gave product 30. Subsequent carbonylation and removal of the N-t-Bu group led to spirocyclic carboxylic acid 31, which was subsequently coupled with aminopiperidinone salt 32 in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) to form ubrogepant (Scheme 4).
Siponimod (BAF-312, 37) is a sphingosine-1-phosphate (S1P) receptor modulator developed by Novartis. Sphingosine-1-phosphate regulates various physiological processes (e.g., lymphocyte recirculation and cardiac function), mostly via five G-proteincoupled S1P receptor subtypes. Influencing the behavior of the immune system through modulation of these receptors was promising for the treatment of autoimmune diseases, and the approval of fingolimod (33) for the treatment of the relapsing form of multiple sclerosis verified this concept. Fingolimod is phosphorylated in vivo by sphingosine kinase, and the resulting active metabolite fingolimod phosphate binds with nanomolar affinity as an agonist at S1P1, S1P3, S1P4, and S1P5 receptors. Then, it induces rapid and persistent internalization of the S1P1 receptor what is required for lymphocyte egress from the secondary lymphoid organs. Overall, it acts as a functional S1P1 receptor antagonist. Unfortunately, treatment with fingolimod causes transient, dosedependent decrease of heart rate (bradycardia). In mice, this side effect was mostly connected to S1P3 receptors. As a result, attempts were made to develop selective S1P1 receptor modulators (Fig. 4) [25, 26].
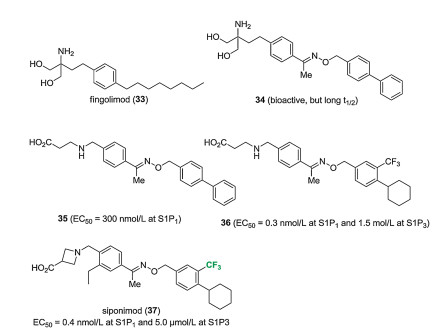
The first step in siponimod development was to increase the rigidity of the hydrophobic octyl chain of fingolimod. Benzyloxy imine 34 retained the bioactivity of 33, but its phosphate had very long in vivo elimination half-time (t1/2> 30 h). To reduce t1/2 and avoid the necessity of activation by sphingosine kinase, the amino alcohol head was replaced with an amino acid part. Compound 35, one of the most effective of the resulting compounds (EC50 = 300 nmol/L at S1P1), was chosen for further optimization. In the case of the biphenyl part, the introduction of a CF3 group on the phenylene ring in ortho position to phenyl and replacement of the phenyl group with cyclohexyl greatly improved S1P1 affinity, while S1P3 affinity was not much affected (compound 36, EC50 = 0.3 nmol/L at S1P1 and 1.5 μmol/L at S1P3). Finally, optimization of the other side of the molecule was achieved by the introduction of an Et group on the benzene ring to decrease S1P3 affinity and replacement of the β-alanine part with an azetidine carboxylic acid part to improve S1P1 affinity. The result was BAF-312 (37, EC50 = 0.4 nmol/L at S1P1 and 5.0 μmol/L at S1P3), which was sent to clinical trials in patients with different autoimmune diseases [26].
Although siponimod has S1P1/S1P5 selectivity (EC50 values: 0.39 nmol/L at S1P1, >10, 000 nmol/L at S1P2, >1000 nmol/L at S1P3, 750 nmol/L at S1P4, and 0.98 nmol/L at S1P5), it still causes bradycardia at treatment onset [25]. Fortunately, this can be attenuated by dose titration [27, 28]. Later findings suggest that in rats (and possibly humans), S1P1 receptors mediate bradycardia, while hypertension (another side effect of fingolimod) is mediated by S1P3 receptor activation [29]. With dose titration, the safety and tolerability profile of siponimod was acceptable. Phase II trials of siponimod in polymyositis and/or dermatomyositis (NCT01801917, NCT02029274, NCT01148810) have been terminated, but trials in sclerosis multiplex were successful, and the FDA approved siponimodin March 2019 to treat adults with relapsing forms of multiple sclerosis (MS), including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease (SPMS) [30]. Previously, no treatment has consistently shown efficacy in slowing disability progression in patients with SPMS [31].
Meanwhile, direct effects of siponimodon the central nervous system were uncovered. This drug can cross the blood–brain barrier, affecting neurobiological processes through modulation of S1P1 on astrocytes and S1P5 on oligodendrocytes [32]. In experimental autoimmune encephalomyelitis, siponimod showed neuroprotective effects (attenuation of astrogliosis and microgliosis, reduced lymphocyte infiltration) [33]. Siponimod attenuated both inflammatory and non-inflammatory models of demyelination in organotypic slice cultures (on oligodendrocytes, S1P5 is expressed and plays a role in myelination) [34]. In animal studies, selective S1P1 functional antagonists like siponimod suppressed intracranial aneurysm through promoting endothelial integrity and blocking macrophage transmigration [35] and attenuated (or even reversed) neuropathic pain [36]. In mice, siponimod attenuated perihemorrhagic edema and improved survival in experimental intracerebral hemorrhage [37]. A Phase II trial (NCT03338998) to investigate the efficacy, safety and tolerability of siponimod in patients with intracerebral haemorrhage is currently in progress [30].
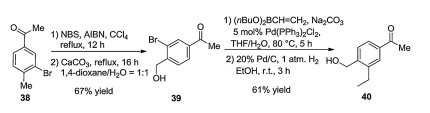
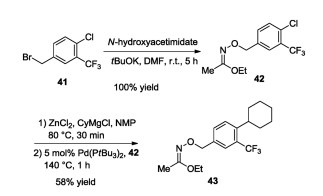
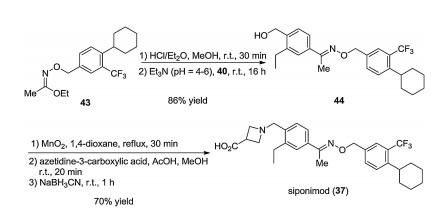
The synthesis of siponimod (37), its salts, and polymorphs was described first in various patents [38-41]. Later, Novartis disclosed a detailed synthetic pathway. In this process, one part of the molecule was obtained from 3-bromo-4-methylacetophenone 38 by hydroxylating the methyl function attached to the benzene ring, exchange of the bromine atom with a vinyl group via Suzuki crosscoupling followed by saturation of the introduced vinyl group. The other part was synthesized by O-alkylating the N-hydroxyacetimidate moiety with 4-(bromomethyl)-1-chloro-2-(trifluoromethyl)benzene (41) to obtain aryl chloride 42, followed by Negishi cross-coupling with cyclohexyl zinc chloride prepared in situ. Then, the iminoether moiety of compound 43 was hydrolyzed, followed by condensation with compound 40. The hydroxymethyl group of compound 44 thus obtained was oxidized to aldehyde, followed by reductive amination with azetidine-3-carboxylic acid to obtains iponimod (37) (Schemes 5–7) [26]. Subsequently, Novartis patented an improved synthetic pathway [42].
Lemborexant (E2006, 48) was developed by Eisai, a Japanese pharmaceutical company. Its FDA approval for the treatment of insomnia characterized by difficulties with sleep onset and/or sleep maintenance in adults was granted in December 2019.
Orexin A and orexin B are endogenous neuropeptides with receptors (OX1 and OX2) expressed throughout the central nervous system. The orexin pathway plays an important role in regulating the sleep/wake cycle, and the need to develop sleep medications with new mechanisms of action directed attention to it. However, it was not clear if a dual receptor antagonist or an OX2 receptor selective antagonist will be a superior sleep aid. Research within Eisai towards orally active orexin receptor antagonists resulted in a compound family with a cyclopropane core [43].
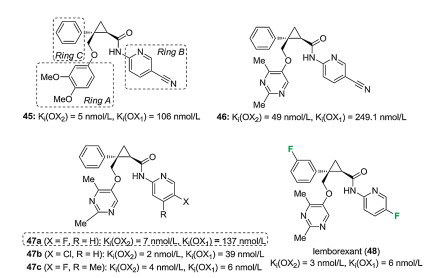
Of the synthesized molecules, 45 was selected for further optimization. This compound had a good affinity for the OX2 receptor (Ki = 5 nmol/L at OX2 and 106 nmol/L at OX1), but had low solubility (both at pH 1.2 and pH 7.4), and showed timeindependent inhibition (TDI) of CYP3A as well as moderate reversible inhibition of other CYPs. Optimization started with Ring A (Fig. 5). It was suspected that O-demethylation and subsequent oxidation to reactive quinones may be behind the TDI of CYP3A. To avoid this, while preserving the shape of the molecule and improve its solubility, alkylated heterocycles were introduced as Ring A. Of the synthesized analogues, compound 46 was amongst the best in terms of solubility and TDI of CYP3A. This compound was chosen for further optimization. Compared to other derivatives of 45 modified in Ring A, compound 46 had a lower affinity for OX receptors (Ki = 49 nmol/L at OX2 and 2.491 μmol/L at OX1). Modifications of Ring B focused on alterations without adversely affecting other parameters. Highly active compounds were obtained with Ring B variants bearing 5-fluoropyridin-2-yl (47a, Ki = 7 nmol/L at OX2 and 137 nmol/L at OX1), 5-chloropyridin-2-yl (47b, Ki = 2 nmol/L at OX2 and 39 nmol/L at OX1) or 5-fluoro-4- methylpyridin-2-yl (47c, Ki = 4 nmol/L at OX2 and 6 nmol/L at OX1) substituents. However, 47c had decreased solubility. From the other two, 47a was optimized further because of its well-balanced in vitro profiles. This optimization was a fluorine scan on Ring C (Fig. 5). From the prepared molecules, lemborexant (48, Ki = 3 nmol/L at OX2 and 6 nmol/L at OX1) showed the most promising balance of in vitro properties. Further preclinical studies showed very weak reversible inhibition of CYP, weak TDI effects on CYP3A, and sufficient brain penetration. As a consequence, lemborexant was sent to further preclinical and clinical studies [44].
In mice experiments, lemborexant (48) caused a dose-dependent increase of REM sleep time while compound 45 did not provoke REM-sleep even up to the highest dose. This strongly indicated that both receptors should be targeted to promote physiological sleep by increasing both non-REM and REM sleep [44]. A plasma concentration–response (CR) modeling method of robust ECG data from Phase I studies proved that lemborexant will not cause clinically concerning QT prolongation, obviating the need for a thorough QT study [45]. A Phase II study demonstrated that lemborexantis well tolerated, has an acceptable safety profile, and it is efficient for the treatment of insomnia, while minimizing next-morning residual sleepiness [46]. Phase III data reinforced these findings [47] and the drug was approved by the FDA in December 2019.
A synthesis of lemborexant (48) was published together with details of its development and data of initial animal studies. The first step is the reaction of 2-(3-fluorophenyl)acetonitrile with (R)- epichlorohydrin to form lactone 50 condensed with cyclopropane. After reductive lactone opening, enzyme-catalyzed regioselective acylation yielded intermediate 52. With its unprotected hydroxymethyl group, compound 52 was capable of alkylating 2, 4- dimethyl-pyrimidine-5-ol under Mitsunobu conditions with the use of diisopropyl azodiformate (DIAD), and subsequent deacetylation yielded derivative 53. The primary alcohol function of 53 was oxidized to carboxylic acid 54, which was then subjected to O-(7- azabenzotriazol-1-yl)-N, N, N', N'-tetramethyluronium hexafluorophosphate (HATU)-mediated coupling with 2-amino-5-fluoropyridine to deliver lemborexant (48) (Scheme 8) [44].
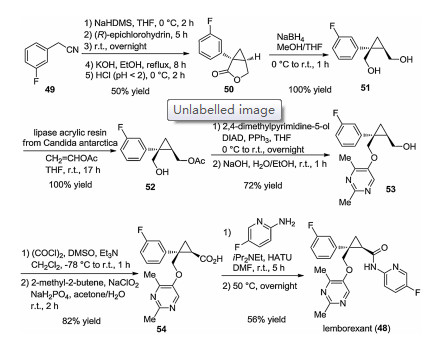
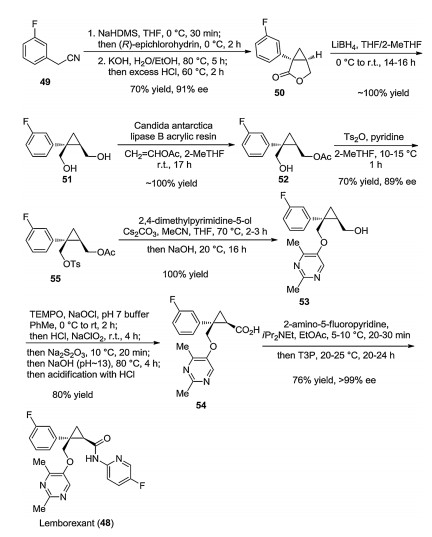
Eisai described a variant of the above synthetic pathway in a patent published in 2013 [48]. This seems to be more suited for larger-scale lemborexant production. For most products, only their mass values were given, but these allow easy calculation of yields. This pathway is shown below on Scheme 9.
Upadacitinib (ABT-494, 56A) was discovered by Abbott Laboratories and developed by its spin-off company, AbbVie Inc. It was approved by the FDA in August 2019 for the treatment of adults with rheumatoid arthritis (RA) with moderate to severe activity, who have had an inadequate response or intolerance to methotrexate (Fig. 6). Abbott Laboratories also did a thorough structural scan based on the key tricyclic core. Systematic variation of the heterocyclic cores, the side chain and the amido moiety, led to upadacitinib (56A) featuring a 2, 2, 2-trifluoroethyl on the nitrogen atom [51].
Rheumatoid arthritis is a systemic autoimmune disease characterized by chronic inflammatory synovitis and progressive joint destruction. These pathological processes involve inflammatory cytokines and auto-reactive T cells. Janus kinases have a key role in triggering the symptoms of rheumatoid arthritis and other autoimmune diseases, because their phosphorylation is often part of the cytokine-activated cell signaling pathways. There are four JAKs (JAK1, JAK2, JAK3 and TYK2) and each has highly specific functions in the control of various immune responses. Taking these into account, numerous JAK inhibitors with different selectivities were synthesized and sent to clinical trials, including upadacitinib [49, 50]. This molecule is a selective JAK1 inhibitor (IC50 for JAK1: 0.043 μmol/L; JAK2: 0.2 μmol/L; JAK3: 2.3 μmol/L; TYK2: 4.7 μmol/L), which showed efficacy, safety, and tolerability in clinical trials of patients with various autoimmune diseases.
In the case of rheumatoid arthritis, after the successful Phase II trials, a number of Phase III trials were completed for patients with inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs or refractory to biologic disease-modifying anti-rheumatic drugs [52, 53]. In all cases, upadacitinib exhibited safety (although higher doses slightly increased the risk of infections) and efficacy. In fact, another Phase III trial found that efficacy of upadacitinib was superior to that of adalimumab [54]. These led to FDA approval of upadacitinib in August 2019.
In subjects with moderate to severely active Crohn's disease, who had responded inadequately to or were intolerant to immunomodulators or anti-TNF therapy, a Phase II trial proved the induction of symptomatic and endoscopic remission [55]. Safety was similar to other studies of upadacitinib. Phase III studies are still ongoing [56]. A Phase II study of upadacitinib in patients with ulcerative colitis showed a dose response [57].
Treatment of patients with active ankylosing spondylitis, who had an inadequate response or contraindication to non-steroidal anti-inflammatory drugs (Phase II/III trial SELECT-AXIS 1) showed that 15 mg of upadacitinib was efficacious and well tolerated. No serious infections, herpes zoster, malignancy, venous thromboembolic events, or deaths were reported. A Phase III study (NCT04169373) is currently in the recruitment phase.
In adults with moderate to severe atopic dermatitis, upadacitinib was efficacious (NCT02925117) and appeared to exhibit a favorable benefit/risk profile (infections were more common with upadacitinib than with placebo, but only a few were serious) [58]. Phase III studies are also ongoing in psoriatric arthritis and giant cell arteritis.
Synthetic pathways towards upadacitinib are only found in patents. The patent of Abbott Laboratories, which covered the structure of upadacitinib first contains information about the synthesis, but since substeps are described in distant parts of the document, reconstruction of the synthetic pathway is difficult [59]. A patent filed by AbbVie slightly later is more user-friendly, but its loose descriptions (e.g., preferably ambient temperature to about 60 ℃) make it difficult to reproduce [60]. A 2017 AbbVie patent, however, contains a synthetic pathway described in detail. First, compound 57 is used to arylate ethyl carbamate. Then the pyrrolidine-containing part of the molecule is synthesized from Cbz-protected glycine ester 59. Compound 59 is reacted with ethyl acrylate (conjugate addition, then intramolecular Claisen condensation) to form highly enolized ester intermediate 60. After O-sulfonylation, an ethyl group is introduced by Suzuki crosscoupling, yielding 62. After ester hydrolysis, enantioselective hydrogenation and addition of dicyclohexylamine results in salt 64 (Schemes 10 and 11).
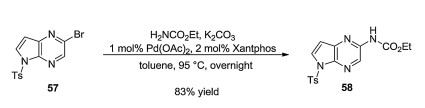
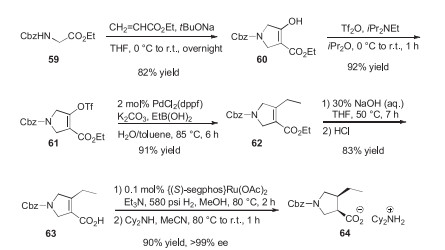
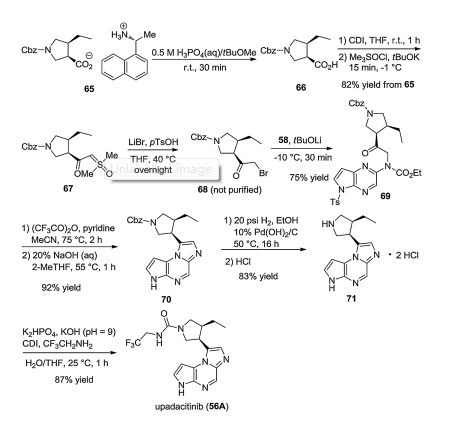
Interestingly, the synthesis is continued with (R)-1-(naphthalen-l-yl)ethanamine salt 65 (64 and 65 have the same carboxylate anion). From 65, pyrrolidine carboxylic acid 66 is regenerated and its COOH group is transformed into COCH2Br group with the help of sulfoxonium ylides. Resulting compound 68 is used to N-alkylate the carbamate moiety of 58. This is followed by cyclization with trifluoroacetic anhydride and Cbz deprotection (hydrogenolysis). Finally, CDI and 2, 2, 2-trifluoroethylamine [61] are used to create the carbamide function [62]. Alternative synthesis pathways can be found in a 2019 patent filed by Dr. Reddy's Laboratories (Scheme 12) [63].
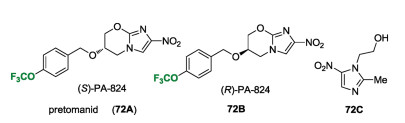
Pretomanid ((S)-PA-824) (72A) was developed by the Global Alliance for TB Drug Development (TB Alliance) as a most promising anti-tuberculosis drug candidate [64-66]. Pretomanid (72A) features two key moieties, bicyclic nitroimidazole and trifluoromethoxy substituted benzyl ether with an (S) absolute configuration carbon center (Fig. 7). Actually, other related compounds, such as metronidazole (72C) and OPC-67683, have been reported as the potential nitroimidazole drug for tuberculosis. However, PA-824 (72) has been demonstrated to be the promising anti-TB drug candidate with IC50 values of 0.015- 0.25 μmol/L [66a]. The in vitro anti-TB activity of 72A was evaluated against H37Rv Mtb in GAST media and 7H12 media with IC50 value of 0.12 μmol/L and 0.09 μmol/L respectively [66b]. It should be mentioned that (S)-PA-824 (72A) has antibacterial activity against Mycobacterium tuberculosis in vitro and used in phase clinical trials for tuberculosis (TB). In sharp contrary, (R)-PA- 824 (72B) is inactive to Mycobacterium tuberculosis [65]. It gets its first approval from FDA in 2019 for the treatment of adults with drug resistant tuberculosis [67].
A four-step method for the synthesis of pretomanid (72A) was presented in Scheme 13 [66a], which was developed by Reider, Sorensen, and co-authors in 2010. Esterification reaction of (R)-3- chloro-1, 2-propanediol (73) with p-methoxybenzoyl chloride in dichloromethane with imidazole as the base afforded the ester 74 in 78% yield and >99% ee. On the other hand, treatment of 4- (trifluoromethoxy)benzyl alcohol by NaH in methyl t-butyl ether at room temperature followed by addition reaction to trichloroacetonitrile at 0 ℃ led to the formation of 4-(trifluoromethoxy)benzyl trichloroacetimidate (75) in 98% yield. In the presence of stoichiometric TsOH, the trichloroacetimidate intermediate (75) was converted into glycidol-derived alkyl chloride 76 with 80% yield after stirring at room temperature. Then, the substitution reaction of intermediate 76 by 2-chloro-4-nitroimidazole in the presence of sodium iodide and potassium carbonate in DMF at 120 ℃ gave the intermediate 77. Finally, deprotection of p-methoxybenzoyl group and intramolecular cyclization was accomplished by exposing to the basic conditions affording pretomanid (72A) in 62% yield, which was recrystallized from the solvent of iPrOH/hexane giving the pure product in 99.9% ee. This method is a significantly improved strategy comparing with the original synthesis with 2, 4-dinitroimidazole as the starting material [68], and also could be used for large-scale preparation.

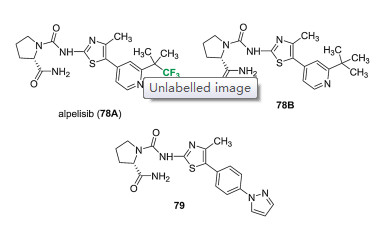
Alpelisib (NVP-BYL719, 78A) is a potent phosphatidylinositol-3 kinase α (PI3Kα) inhibitor, which was developed by Novartis for the treatment of breast cancer (Fig. 8) [69-71]. The 2-aminothiazole scaffold has been found to be a valuable template for the development of highly selective PI3K inhibitors, in particular, with a urea linked (S)-pyrrolidinecarboxamide unit to the 2-amino group (79). Alpelisib (78A) is also a chiral 2-aminothiazole derivative, and the (S)-pyrrolidine carboxamide moiety linked to aminothiazole unit via a urea bond was optimized to be the best choice in this class of PI3K inhibitors. The existence of trifluoromethyl group was also found to produce a potent double digit nanomolar inhibition of PI3Kα-dependent Akt activation, comparing to the compound 78B featuring a tert-butyl group. In the biochemical assay of the inhibition of p110α, p110β, p110δ and p110γ activity, the IC50 values (p110α, p110β, p110δ and p110γ) for alpelisib (78A) were 0.005, 1.2, 0.29 and 0.25 μmol/L, respectively. While the values for compound 78B were 0.014, 4.4, 0.33 and 0.43 μmol/L, respectively. During the assessment of the metabolic stability via incubation of compounds 78 with rat liver microsomes, alpelisib (78A) containing a trifluoromethyl substituent showed a dramatically reduced in vitro clearance compared to compound 78B. As the IC50 values shown above, it should be mentioned that alpelisib (78A) had a good activity against wildtype PI3Kα, but had an obviously less activity against PI3Kδ, PI3Kγ and PI3Kβ [69, 70]. In 2019, alpelisib was approved by FDA with the trade name of PiqrayTM for treating HR+, HER2-negative, PIK3CAmutated advanced or metastatic breast cancer in combination with fulvestrant [72].
Alpelisib (78A) was accessed as showed in Scheme 14 [69], with the Pd-catalyzed coupling reaction between 4-methyl-2-acetaminothiazole (85) and a 4-bromopyridine intermediate 84 as the key step of this synthetic route. Initially, 3, 3, 3-trifluoro-2, 2-dimethylpropanoic acid was converted into chloride 80 in quantitative yield via the treatment of oxalyl chloride. The resulting chloride 80 was transferred into 4H-pyran-4-one intermediate 82 through the reaction of (E)-4-methoxybut-3-en-2-one (81) in the presence of LiHMDS at -78 ℃ followed by a TFA-mediated cyclization. Amination substitution reaction of 4H-pyran-4-one intermediate 82 using ammonia at 65 ℃ afforded pyridin-4(1H)-one 83, which was treated by POBr3 to give the bromide intermediate 84 in 51% yield. Then, Pd-catalyzed coupling reaction between intermediate 84 and 4-methyl-2-acetaminothiazole (85) gave the intermediate 86, which was subjected to the hydrolysis reaction in the presence of 6 mol/L HCl. The condensation reaction of the obtained amine 87 with 1H-imidazole-1-carboxylic acid generated the imidazole intermediate 88, which was substituted by (S)-pyrrolidine-2- carboxamide (89) at room temperature to afford alpelisib (78A).

Selinexor (KPT-330) (90A) has the chemical structure as shown in Fig. 9, which was developed by Therapeutics as a first-in-class selective inhibitor of Exportin-1 (XPO1) for the treatment of cancer. Selinexor contains three key moieties, including trifluoromethylated phenyl, 1, 2, 4-triazole and 2-hydrazinylpyrazine. Actually, Therapeutics reported a series of hydrazide-based nuclear transport modulators, which featured a bis-trifluoromethylated phenyl key structural unit in all these compounds (90A-C). The compound 90A also was examined in various breast cancer cell lines, and low IC50 values were found (0.01 μmol/L for MDA-MB- 486, 0.01 μmol/L for MDA-MB-231, and 0.013 μmol/L for DU4475). Selinexor (90A) showed good cytotoxicity in a wide range of myeloid leukaemia cell lines with less than 0.5 μmol/L of IC50 values. Furthermore, synergistic anticancer activity was observed in preclinical studies, when selinexor (90A) was used as a combination with other drugs. In 2019, selinexor gets its first global approval by FDA with the trade name of XpovioTM for treating adults with relapsed or refractory multiple myeloma (RRMM) [73, 74].
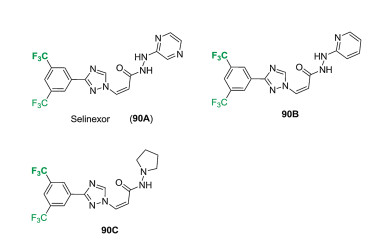
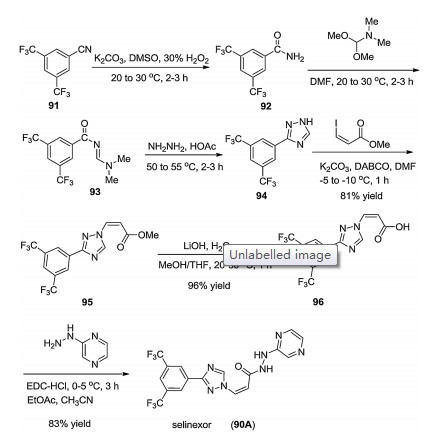
The procedure for the process of selinexor (90A) was outlined in Scheme 15, starting from 3, 5-bis(trifluoromethyl)benzonitrile. Hydrolysis of benzonitrile 91 by the treatment of potassium carbonate in the presence of 30% H2O2 provided the 3, 5-bis (trifluoromethyl)benzamide (92), which was subjected to the reaction with N, N-dimethyl formamide dimethyl acetal affording the imine intermediate 93. Subsequent HOAc-catalyzed cyclization reaction of imine intermediate 93 with hydrazine at 50-55 ℃ generated 1, 2, 4-triazole intermediate 94. Substitution of (Z)- methyl 3-iodoacrylate by the intermediate 94 in DMF by the use of base gave ester 95 in 81% yield, which was transferred into acid 96 by treating of LiOH. Finally, the coupling reaction between acid 96 and 2-hydrazinylpyrazine in the presence of 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide hydrochloride (EDC-HCl) afforded selinexor (90A) in 83% yield [75].
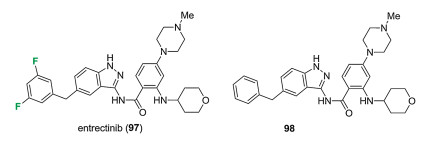
Entrectinib (97) was discovered by Nerviano Medical Sciences as a potent anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 kinase (ROS1) inhibitor [76-79]. Entrectinib (97) features the core structure of 3-aminoindazole, and other key moieties including fluorinated benzyl, acyl and piperazine groups (Fig. 10) [76]. In particular, the two fluorine substitutions were found to be very important for the biochemical activity via the structure-activity relationship (SAR) study. For example, compound 98 without fluorine substitution shows about 10-fold decrease of biochemical activity on ALK (IC50 = 0.106 μmol/L vs. 0.012 μmol/L for fluorinated compound 97) and also decrease of antiproliferative activity in Karpas-299 cell line (IC50 = 0.182 μmol/L vs. 0.031 μmol/L for fluorinated compound 97). Entrectinib (97) was also found to be very efficient as the inhibitor for the growth of TRKA-, TRKB-, and TRKC-dependent Ba/F3 cell lines, and low IC50 values were found (0.003 μmol/L for each cell line). Entrectinib (97) was firstly approved by Japan in June of 2019. Then in August of 2019, it got its approval by FDA with the trade name RozlytrekTM for treating ROS1-positive non-small cell lung cancer and NTRK gene fusion-positive solid tumors [80].
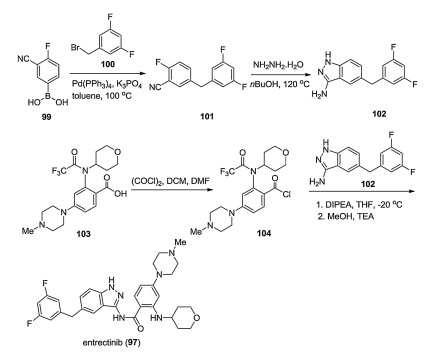
Nerviano also developed a synthetic method for the synthesis of entrectinib (97) (Scheme 16) [76], which started from (3-cyano-4- fluorophenyl)boronic acid (99). Pd-catalyzed coupling reaction between boronic acid 99 and benzyl bromide 100 in toluene at 100 ℃ yielded diphenyl methane 101, which underwent the cyclization reaction with hydrazine hydrate in butanol under reflux resulting in the key 3-aminoindazole intermediate 102. Subsequently, compound 102 was subjected to the amidation reaction with acyl chloride 104 in the presence of DIPEA at -20 ℃ leading to the final entrectinib (97).
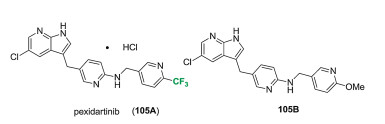
Pexidartinib (105A) was discovered by Daiichi Sankyo, and used as a colony-stimulating factor 1 (CSF1) inhibitor for the treatment of symptomatic tenosynovial giant cell tumor (TGCT). The chemical structure of pexidartinib (105A) was shown in Fig. 11, which features a trifluoromethyl pyridine moiety. Daiichi Sankyo Company developed a process for the preparation of pexidartinib (105A) in 2008. In 2016, they patented a series of related compounds with activities on the receptor protein tyrosine kinase c-KIT and c-FMS [81, 82]. Several kinds of substitutions on the core heterocyclic moieties, including non-fluorinated group (105B), were examined. Pexidartinib (105A) showed high selectivity as the inhibitor of CSF1R and KIT receptor tyrosine kinases, with the IC50 value as 0.02 μmol/L and 0.01 μmol/L respectively [83]. Pexidartinib was approved by FDA in August of 2019 for treating the disease of TGCT associated with severe morbidity or functional limitations and not amenable to Surgery [83].
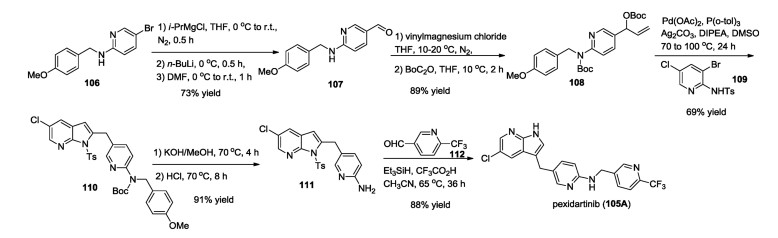
The synthetic method including large-scale synthesis for pexidartinib (105A) was developed by Shanghai Institute of Pharmaceutical Industry in 2019 [84], which started from 5- bromo-N-(4-methoxybenzyl)pyridin-2-amine (106) via a key tandem Tsuji–Trost reaction and Heck coupling (Scheme 17). The reaction with DMF in the presence of BuLi leading to the aldehyde 107 in 73% yield. Then, aldol reaction between aldehyde 107 and vinylmagnesium chloride, followed by the Boc protection resulted in the intermediate 108. Pd-catalyzed tandem Tsuji–Trost reaction and Heck coupling between intermediate 108 and pyridine intermediate 109 gave the key 1H-pyrrolo[2, 3-b]pyridine intermediate 110 in 69% yield. Deprotection of Boc and PMB groups afforded the free amine 111, which was finally converted into pexidartinib (105A) via a reduction amination reaction. It should be mentioned that Daiichi Sankyo Company also reported the synthetic methods for the preparation of pexidartinib (105A) in 2008 and 2016, which used 5-chloro-1H-pyrrolo[2, 3-b]pyridine as the key reagent [81, 82].
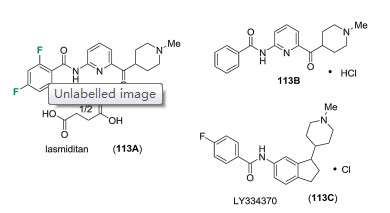
Lasmiditan (113A), also known as COL-144 and LY573144, was developed by Eli Lilly, as a highly selective serotonin 5- hydroxytryptamine (5-HT)1F receptor agonist. Lasmiditan (113A) is a succinic acid salt, which features 2-amino-6-(piperidin-4- carbonyl)pyridine as the key structural core (Fig. 12). Eli Lilly also carried out a systematic study on scanning the structure of the heterocyclic and the amido groups, which led to the compound 113A featuring 2, 4, 6-trifluorinated benzoyl moiety with improved pharmacological properties [85-89]. Lasmiditan (113A) was different from the early prototype indole-based 5-HT1F receptor agonist, LY334370 (113C). In binding studies, lasmiditan (113A) showed excellent selectivity at human 5-HT receptor. The Ki value at 5-HT1F receptor was 2.2 nmol/L, while dramatically increased Ki values (470-fold) at 5-HT1A, 5-HT1B, and 5-HT1D receptor were found as 1053 nmol/L, 1043 nmol/L and 1357 nmol/L respectively [85]. In October of 2019, lasmiditan (113A) was approved by FAD with the trade name ReyvowTM for treating migraine with or without aura in adults [88].
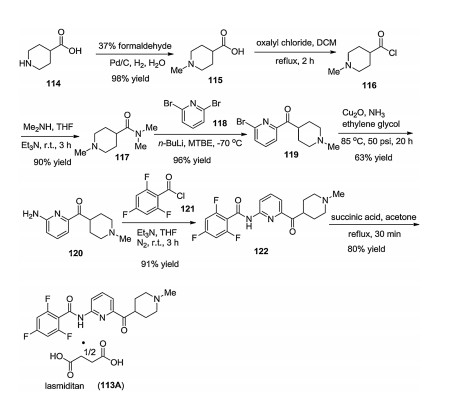
The procedure for the process of lasmiditan (113A) was presented in Scheme 18 [89]. Reductive methylation on the nitrogen atom of isonipicotic acid (114) by the use of formaldehyde in the presence of Pd/C catalyst gave acid 115, which was converted into acyl chloride 116 by reflux with oxalyl chloride in DCM for 2 h. Addition of dimethylamine into the THF solution of chloride 116 at room temperature in the presence of triethylamine afforded the amide 117 in 90% yield. On the other hand, treating 2, 6- dibromopyridine (118) by butyl lithium at -70 ℃ gave the lithium salt, which then reacted with amide 117 in methyl t-butyl ether affording the intermediate 119. Then, Cu-catalyzed coupling reaction between NH3 and bromide intermediate 119 in pressure autoclave at 85 ℃ for 20 h generated amine 120 in 63% yield. Amidation reaction between amine 120 and 2, 4, 6-trifluorobenzoyl chloride (121) in THF with triethylamine as a base afforded 122 in 91% yield. Finally, treatment of compound 122 with 0.5 equiv. of succinic acid in acetone under reflux for 30 min afforded lasmiditan (113A)
The eleven new fluorinated drugs discussed in this review article feature aromatic fluorination inlumateperone (4A), lemborexant (48), entrectinib (97), lasmiditan (113A); aromatic CF3 groups in siponimod (37), selinexor (90A), pexidartinib (105A); aliphatic CF3 groups in ubrogepant (14A), upadacitinib (56A), alpelisib (78A); and finally, a CF3O group in pretomanid (72A). Similar to the general trend observed in the previous years [6], the aromatic fluorination (one, two or three fluorine atoms on an aromatic ring) continues to be a dominant type of fluorination. However, the relative number of compounds containing aromatic and aliphatic trifluoromethyl group has noticeably increased. In particular, we would like to point out ubrogepant (14A) and upadacitinib (56A) bearing pharmacophoric CF3CH2N moiety. In general, the trifluoromethyl group is becoming an increasingly common trait among marketed pharmaceuticals and drugcandidates [6c, 90]. Considering the progress in the synthetic methodology of trifluoromethylation [91], one may expect this trend will continue to grow.
Another trend one can observe in the compounds under discussion is that over 50% of new drugs contain at least one stereogenic center and were approved to be administered in enantiomerically pure form. In this regard, we would like to highlight the importance of the phenomenon of Self-Disproportionation of Enantiomers (SDE). In particular, fluorine is an established SDE-phoric structural element [92] due tothepropensityoffluorinecontaining compounds to exhibit SDE more commonly and with an enhancedmagnitudeincomparison tonon-fluorinatedcounterparts [93]. The role that fluorine plays is central in many extreme cases of the SDE, particularly those involving achiral chromatography [94] and sublimation [95]. Furthermore, the necessity to habitually perform SDE tests to ensure the accuracy of the enantiomeric composition of materials obtained from catalytic asymmetric transpositions should be ever strongly emphasized [96]. The potential consequences of ignoring the SDE phenomenon are significant errors in reporting the stereochemical outcome of catalytic asymmetric transformations in terms of the enantiomeric composition of the catalysts and products as well as peculiarities in the properties of chiral pharmaceuticals. Since there are a rapidly growingnumberoffluorine-containingdrugson thepharmaceutical market, the necessity for regulations with a clear understanding of the SDE properties for the safe handling, production, processing, storage, and medical applications of fluorine-containing drugs is paramount.
Another imperative trend is the increasing number (~73%) of drugs containing both fluorine and a motive of tailor-made amino acid. The inclusion of an amino acid residue in drug-molecules allows to improve both functional and structural complexity by providing two orthogonal functionalities as well as a stereogenic center [97]. These factors usually bode well for the success rate of drug-candidates. Of particular significance is that promiscuity and off-target toxicity are known to be lower with the increasing number of stereogenic centers, further underscoring the pharmaceutical potential of chiral compounds and the importance of SDE phenomenon.
As the final note of caution, we would like to point out the inacceptable lack of study into metabolism of fluorinated pharmaceuticals. To guarantee an effective progress in the appreciation of fluorine in healthcare industry, the emerging concern over an overload of fluoride, as a possible final metabolite of organo-fluorine drugs, should be properly addressed [98].
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21761132021) and IKERBASQUE, Basque Foundation for Science. The Hungarian Research Foundation (NKFIH No. K119282) and Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT are also acknowledged.
(a) J. Fried, E.F. Sabo, J. Am. Chem. Soc. 75 (1953) 2273-2274;
(b) J. Fried, E.F. Sabo, J. Am. Chem. Soc. 76 (1954) 1455-1456.
(a) C. Heidelberger, N.K. Chaudhuri, P. Danneberg, et al., Nature 179 (1957) 663-666;
(b) D.B. Longley, D.P. Harkin, P.G. Johnston, Nat. Rev. Cancer 3 (2003) 330-338.
D. O'Hagan, J. Fluorine Chem. 131 (2010) 1071-1081. doi: 10.1016/j.jfluchem.2010.03.003
J.A. Tobert, Nat. Rev. Drug Discov. 2 (2003) 517-526. doi: 10.1038/nrd1112
(a) Q.A. Huchet, B. Kuhn, B. Wagner, et al., J. Med. Chem. 58 (2015) 9041-9060;
(b) B. Jeffries, Z. Wang, H.R. Felstead, et al., J. Med. Chem. 63 (2020) 1002-1031;
(c) A.D. Wade, A. Rizzi, Y. Wang, D.J. Huggins, J. Chem. Inf. Model. 59 (2019) 2776-2784;
(d) E.N.G. Marsh, Acc. Chem. Res. 47 (2014) 2878-2886;
(e) A.A. Berger, J.S. Völler, N. Budisa, B. Koksch, Acc. Chem. Res. 50 (2017) 2093-2103;
(f) L. Kiss, A.M. Remete, Eur. J. Org. Chem. (2019) 5574-5602;
(g) L. Kiss, F. Fülöp, Chem. Rec. (2018) 266-281;
(h) A.M. Remete, M. Nonn, S. Fustero, F. Fülöp, L. Kiss, Tetrahedron 74 (2018) 6367-6418;
(i) M. Nonn, L. Kiss, M. Haukka, S. Fustero, F. Fülöp, Org. Lett. 17 (2015) 1074-1077;
(j) J. Liu, Z. Li, H. Mei, V.A. Soloshonok, J.L. Han, ACS Omega 4 (2019) 19505-19512;
(k) C. Xie, L. Zhang, W. Sha, et al., Org. Lett. 18 (2016) 3270-3273;
(l) X. Xu, X. Dong, Z. Zhang, et al., Macroheterocycles 12 (2019) 403-408.
(a) J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432-2506;
(b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J.L. Aceña, V.A. Soloshonok, K. Izawa, H. Liu, Chem. Rev. 116 (2016) 422-518;
(c) W. Zhu, J. Wang, S. Wang, et al., J. Fluorine Chem. 167 (2014) 37-54;
(d) Y. Zhu, J.L. Han, J. Wang, et al., Chem. Rev. 118 (2018) 3887-3964;
(e) H. Mei, J. Han, K.D. Klika, et al., Eur. J. Med. Chem. 186 (2020) 111826;
(f) N.A. Meanwell, J. Med. Chem. 61 (2018) 5822-5880.
H. Mei, J. Han, S. Fustero, et al., Chem. Eur. J. 25 (2019) 11797-11819. doi: 10.1002/chem.201901840
H.A. Blair, Drugs 80 (2020) 417-423. doi: 10.1007/s40265-020-01271-6
A.J. Robichaud, T. Lee, W. Deng, et al., PCT/US2000/016498, 2000.
T. Lee, A.J. Robichaud, K.E. Boyle, et al., Bioorg. Med. Chem. Lett. 13 (2003) 767-770. doi: 10.1016/S0960-894X(02)01028-4
P. Li, Q. Zhang, A.J. Robichaud, et al., J. Med. Chem. 57 (2014) 2670-2682. doi: 10.1021/jm401958n
R.E. Davis, C.U. Correll, Expert Rev. Neurother. 16 (2016) 601-614. doi: 10.1080/14737175.2016.1174577
N. Zisapel, Expert Opin, Investig. Drugs 24 (2014) 401-411.
J.A. Lieberman, R.E. Davis, C.U. Correll, et al., Biol. Psychiatry 79 (2016) 952-961. doi: 10.1016/j.biopsych.2015.08.026
K.E. Vanover, R.E. Davis, Y. Zhou, et al., Neuropsychopharmacology 44 (2019) 598-605. doi: 10.1038/s41386-018-0251-1
F. Carponi, C. Fabbri, I. Bitter, et al., Eur. Neuropsychopharmacol. 29 (2019) 971-985. doi: 10.1016/j.euroneuro.2019.06.008
I.M. Bell, M.E. Fraley, S.N. Gallicchio, et al., US2010/0122899, 2012.
P.R. Holland, P.J. Goadsby, Neurotherapeutics 15 (2018) 304-312. doi: 10.1007/s13311-018-0617-4
P. Martelletti, M.A. Giamberardino, Expert Opin. Pharmacol. 20 (2018) 209-218.
R.B. Lipton, D.W. Dodick, J. Ailani, et al., JAMA 322 (2019) 1887-1898. doi: 10.1001/jama.2019.16711
I.M. Bell, M.E. Fraley, S.N. Gallicchio, et al., PCT/US2011/060081, 2012.
F. Chen, C. Molinaro, W.P. Wuelfing, et al., PCT/US2013/030692, 2013.
K.M. Belyk, E. Cleator, S.C. Kuo, et al., PCT/US2013/030696, 2013.
N. Yasuda, E. Cleator, B. Kosjek, et al., Org. Process Res. Dev. 21 (2017) 1851-1858. doi: 10.1021/acs.oprd.7b00293
P. Gergely, B. Nuesslein-Hildesheim, D. Guerini, et al., Brit. J. Pharmacol. 167 (2012) 1035-1047. doi: 10.1111/j.1476-5381.2012.02061.x
S. Pan, N.S. Gray, W. Gao, et al., ACS Med. Chem. Lett. 4 (2013) 333-337. doi: 10.1021/ml300396r
E. Legangneux, A. Gardin, D. Johns, Br. J. Clin. Pharmacol. 75 (2012) 831-841.
K. Selmaj, D.K.B. Li, H.P. Hartung, et al., Lancet Neurol. 12 (2013) 756-767. doi: 10.1016/S1474-4422(13)70102-9
R.M. Fryer, A. Muthukumarana, P.C. Harrison, et al., PLoS One 7 (2012) e52985. doi: 10.1371/journal.pone.0052985
Z.T. Al-Salama, Drugs 79 (2019) 1009-1015. doi: 10.1007/s40265-019-01140-x
L. Kappos, A. Bar-Or, B.A.C. Cree, Lancet 391 (2018) 1263-1273. doi: 10.1016/S0140-6736(18)30475-6
U. Glaenzel, Y. Jin, R. Nufer, et al., Drug Metab. Dispos. 46 (2018) 1001-1013. doi: 10.1124/dmd.117.079574
A. Gentile, A. Musella, S. Bullitta, et al., J. Neuroinflamm. 13 (2016) 207. doi: 10.1186/s12974-016-0686-4
C. O'Sullivan, A. Schubart, A.K. Mir, K.K. Dev, J. Neuroinflamm. 13 (2016) 31. doi: 10.1186/s12974-016-0494-x
R. Yamamoto, T. Aoki, H. Koseki, et al., Br. J. Pharmacol. 174 (2017) 2085-2101. doi: 10.1111/bph.13820
Z. Chen, T.M. Doyle, L. Luongo, et al., PNAS 116 (2019) 10557-10562. doi: 10.1073/pnas.1820466116
T. Bobinger, A. Manaenko, P. Burkardt, et al., Stroke 50 (2019) 3246-3254. doi: 10.1161/STROKEAHA.119.027134
S. Pan, W. Gao, N.S. Gray, Y. Mi, Y. Fan, PCT/US2004/015603, 2004.
Y. Liu, D. Papoutsakis, E. Roddy, PCT/US2009/068346, 2010.
L. Ciszewski, M. de la Cruz, P.H. Karpinski, et al., PCT/US2009/068143, 2010.
M. de la Cruz, P.H. Karpinski, Y. Liu, PCT/US2009/068352, 2010.
F. Gallou, J.M. Sedelmeier, C. Vogel, PCT/EP2013/052106, 2013.
Y. Yoshida, T. Terauchi, Y. Naoe, et al., Bioorg. Med. Chem. 22 (2014) 6071-6088. doi: 10.1016/j.bmc.2014.08.034
Y. Yoshida, Y. Naoe, T. Terauchi, et al., J. Med. Chem. 58 (2015) 4648-4664. doi: 10.1021/acs.jmedchem.5b00217
P.J. Murphy, S. Yasuda, K. Nakai, et al., J. Clin. Pharmacol. 57 (2017) 96-104. doi: 10.1002/jcph.785
P. Murphy, M. Moline, D. Mayleben, et al., J. Clin. Sleep Med. 13 (2017) 1289-1299. doi: 10.5664/jcsm.6800
R. Rosenberg, P. Murphy, G. Zammit, et al., JAMA Netw. Open 2 (2019) e1918254. doi: 10.1001/jamanetworkopen.2019.18254
G.A. Moniz, A.Z. Wilcoxen, F. Benayoud, et al., PCT/US2013/026204, 2013.
S. Nakayamada, S. Kubo, S. Iwata, Y. Tanaka, BioDrugs 30 (2016) 407-419. doi: 10.1007/s40259-016-0190-5
M.E.F. Mohamed, H.S. Camp, P. Jiang, et al., Clin. Pharmacokinet. 55 (2016) 1547-1558. doi: 10.1007/s40262-016-0419-y
N. Wishart, M.A. Argiriadi, D.J. Calderwood, et al., WO2011068881, 2011.
(a) G.R. Burmester, J.M. Kremer, F. Van den Bosch, et al., Lancet 391 (2018) 2503-2512;
(b) M.C. Genovese, R. Fleischmann, B. Combe, et al., Lancet 391 (2018) 2513-2524.
J.S. Smolen, A.L. Pangan, P. Emery, et al., Lancet 393 (2019) 2303-2311. doi: 10.1016/S0140-6736(19)30419-2
R. Fleischmann, A.L. Pangan, I.H. Song, et al., Arthritis Rheumatol. 71 (2019) 1788-1800. doi: 10.1002/art.41032
F. D'Amico, G. Fiorino, F. Furfaro, M. Allocca, S. Danese, Expert Opin. Invest. Drugs 27 (2018) 595-599. doi: 10.1080/13543784.2018.1492547
T. Perez-Jeldres, C.J. Tyler, J.D. Boyer, et al., Front. Pharmacol. 10 (2019) 212. doi: 10.3389/fphar.2019.00212
R. Panaccione, G.R. D'Haens, W.J. Sandborn, et al., Gastroenterology 156 (2019) S-170.
E. Guttman-Yassky, D. Thaçi, A.L. Pangan, et al., J. Allergy Clin. Immunol. (2019) 145 (2020) 877-884. doi: 10.1016/j.jaci.2019.11.025
N. Wishart, M.A. Argiriadi, D.J. Calderwood, PCT/US2010/058572, 2011.
J.W. Voss, H.S. Camp, R.J. Padley, PCT/US2014/062145, 2015.
(a)V.A. Soloshonok, H. Ohkura, K.Uneyama, Tetrahedron Lett. 43 (2002) 5449-5452;
(b) V.A. Soloshonok, T. Ono, I.V. Soloshonok, J. Org. Chem. 62 (1997) 7538-7539;
(c) V.A. Soloshonok, V.P. Kukhar, Tetrahedron 52 (1996) 6953-6964.
A. Allian, J. Jayanth, M.E. Mohamed, et al., PCT/US2016/057372, 2017.
S. Oruganti, B. Kandagatla, S. Sen, et al., PCT/IB2018/055368, 2019.
R. Singh, U. Manjunatha, H.I.M. Boshoff, et al., Science 322 (2008) 1392-1395. doi: 10.1126/science.1164571
S. Patterson, S. Wyllie, L. Stojanovski, et al., Antimicrob. Agents Chemother. 57 (2013) 4699-4706. doi: 10.1128/AAC.00722-13
(a) M.A. Marsini, P.J. Reider, E.J. Sorensen, J. Org. Chem. 75 (2010) 7479-7482;
(b) G.C. Moraski, A.G. Oliver, L.D. Markley, et al., Bioorg. Med. Chem. Lett. 24 (2014) 3493-3498.
S.J. Keam, Drug 79 (2019) 1797-1803. doi: 10.1007/s40265-019-01207-9
W.R. Baker, C. Shaopei, E.L. Keeler, US 6087358, 2000.
P. Furet, V. Guagnano, R.A. Fairhurst, et al., Bioorg. Med. Chem. Lett. 23 (2013) 3741-3748. doi: 10.1016/j.bmcl.2013.05.007
M. Gerspacher, R.A. Fairhurst, R. Mah, et al., Bioorg. Med. Chem. Lett. 25 (2015) 3582-3584. doi: 10.1016/j.bmcl.2015.06.077
F. André, E. Ciruelos, G. Rubovszky, et al., N. Engl. J. Med. 380 (2019) 1929-1940. doi: 10.1056/NEJMoa1813904
A. Markham, Drugs 79 (2019) 1249-1253. doi: 10.1007/s40265-019-01161-6
(a) V.P. Sandanayaka, S. Shacham, D. Mccauley, S. Shechter, WO2013019548, 2013;
(b) M. Garg, D. Kanojia, A. Mayakonda, et al., Oncotarget 8 (2017) 7521-7532
Y.Y. Syed, Drugs 79 (2019) 1485-1494. doi: 10.1007/s40265-019-01188-9
A.R. Muthusamy, S.L. Kanniah, A. Ravi, et al., WO2018129227, 2018.
M. Menichincheri, E. Ardini, P. Magnaghi, et al., J. Med. Chem. 59 (2016) 3392-3408. doi: 10.1021/acs.jmedchem.6b00064
D. Liu, M. Offin, S. Harnicar, B.T. Li, A. Drilon, Ther. Clin. Risk Manag. 14 (2018) 1247-1252. doi: 10.2147/TCRM.S147381
R. Iyer, L. Wehrmann, R.L. Golden, et al., Cancer Lett. 372 (2016) 179-186. doi: 10.1016/j.canlet.2016.01.018
G. Wei, R. Patel, C. Walsh, et al., Eur. J. Cancer 69 (2016) S33.
Z.T. AI-Salama, S.J. Keam, Drugs 79 (2019) 1477-1483. doi: 10.1007/s40265-019-01177-y
C. Zhang, J. Zhang, P.N. Ibrahim, et al., WO2008064255, 2008.
P.N. Ibrahim, M. Jin, S. Matsuura, PCT Int. Appl. WO2016179412, 2016.
Y.N. Lamb, Drugs 79 (2019) 1805-1812. doi: 10.1007/s40265-019-01210-0
D. Chen, Y. Zhang, J. Li, Y. Liu, Synthesis 51 (2019) 2564-2571. doi: 10.1055/s-0037-1612421
D.L. Nelson, L.A. Phebus, K.W. Johnson, et al., Cephalalgia 30 (2010) 1159-1169. doi: 10.1177/0333102410370873
M.D. Ferrari, M. Färkkilä, U. Reuter, et al., Cephalalgia 30 (2010) 1170-1178. doi: 10.1177/0333102410375512
Y.J. Jin, X.W. Yang, X.X. Xie, H. Tian, Y.L. Cui, Drugs Clinic 31 (2016) 1300-1303.
Y.N. Lamb, Durgs 79 (2019) 1989-1996.
M.P. Cohen, D.T. Kohlman, S.X. Liang, V. Mancuso, F. Victor, PCT Int. Appl. WO200384949, 2003.
N.A. Meanwell, J. Med. Chem. 54 (2011) 2529-2591. doi: 10.1021/jm1013693
(a) J.A. Ma, D. Cahard, J. Fluorine Chem. 128 (2007) 975-996;
(b) A. Sato, M.V. Ponomarenko, T. Ono, G.V. Röschenthaler, V.A. Soloshonok, Eur. J. Org. Chem. 2019 (2019) 4417-4421;
(c) H. Mei, J.L. Han, S. White, G. Butler, V.A. Soloshonok, J. Fluorine Chem. 227 (2019)109370;
(d) C. Yang, A. Hassanpour, K. Ghorbanpour, S. Abdolmohammadi, E. Vessally, RSC Adv. 9 (2019) 27625-27639;
(e) Z. Zhang, Adv. Synth. Catal. 359 (2017) 372-383;
(f) S.L. Clarke, G.P. McGlacken, Chem. Eur. J. 23 (2017) 1219-1230;
(g) Y. Ouyang, X.H. Xu, F.L. Qing, Angew. Chem. Int. Ed. 57 (2018) 6926-6929;
(h) J. Kalim, T. Duhail, T.N. Le, et al., Chem. Sci. 10 (2019) 10516-10523.
(a) J. Han, O. Kitagawa, A. Wzorek, K.D. Klika, V.A. Soloshonok, Chem. Sci. 9 (2018) 1718-1739;
(b) M.Yasumoto, H.Ueki, V.A.Soloshonok, J.FluorineChem.131 (2010)266-269.
(a) A.E. Sorochinsky, J.L. Aceña, V.A. Soloshonok, Synthesis 45 (2013) 141-152;
(b) M. Yasumoto, H. Ueki, T. Ono, T. Katagiri, V.A. Soloshonok, J. Fluorine Chem. 131 (2010) 535-539.
(a) A.E. Sorochinsky, T. Katagiri, T. Ono, et al., Chirality 25 (2013) 365-368;
(b) T. Nakamura, K. Tateishi, S. Tsukagoshi, et al., Tetrahedron 68 (2012) 4013-4017.
(a) H. Ueki, M. Yasumoto, V.A. Soloshonok, Tetrahedron Asymmetry 21 (2010) 1396-1400;
(b) J. Han, D.J. Nelson, A.E. Sorochinsky, V.A. Soloshonok, Curr. Org. Synth. 8 (2011) 310-317.
(a) C. Xie, L. Wu, J. Han, V.A. Soloshonok, Y. Pan, Angew. Chem. Int. Ed. 54 (2015) 6019-6023;
(b) J. Han, V.A. Soloshonok, K.D. Klika, J. Drabowicz, A. Wzorek, Chem. Soc. Rev. 47 (2018) 1307-1350.
(a) A. Henninot, J.C. Collins, J.M. Nuss, J. Med. Chem. 61 (2018) 1382-1414;
(b) M.A.T. Blaskovich, J. Med. Chem. 59 (2016) 10807-10836;
(c) D.R.W. Hodgson, J.M. Sanderson, Chem. Soc. Rev. 33 (2004) 422-430;
(d) T. Sato, K. Izawa, J.L. Aceña, H. Liu, V.A. Soloshonok, Eur. J. Org. Chem. 2016 (2016) 2757-2774;
(e) V.A. Soloshonok, K. Izawa, Asymmetric synthesis and application of α-amino acids, ACS Symposium Series 1009, Oxford University Press, Oxford, 2009;
(f) S. Wang, Y. Wang, J. Wang, et al., Curr. Pharm. Des. 23 (2017) 4493-4554.
(a) D. Stepec, M. Ponikvar-Svet, Acta Chim. Slovenica 66 (2019) 255-275;
(b) D. Stepec, G. Tavccar, M. Ponikvar-Svet, Environ. Pollut. 248 (2019) 958-964;
(c) M. Ponikvar, V. Stibilj, B. Zemva, Food Chem.103 (2007) 369-374;
(d) A. Koblar, G. Tavcar, M. Ponikvar-Svet, Food Chem.130 (2012) 286-290.

Figure 1 Structures of fluorine-containing drugs fludrocortisone (1), 5-fluorouracil (2) and Lipitor (3).

Figure 3 Structures of ubrogepant (MK-1602, 14A) and its related derivatives (14B and 14C).

Figure 6 Structure of upadacitinib (ABT-494, 56A) and its non-fluorinated derivative (56B).

Figure 11 Structure of pexidartinib (105A) and its related non-fluorinated derivative (105B).

Figure 12 Structures of lasmiditan (113A) and the related derivatives (113B and 113C).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
























