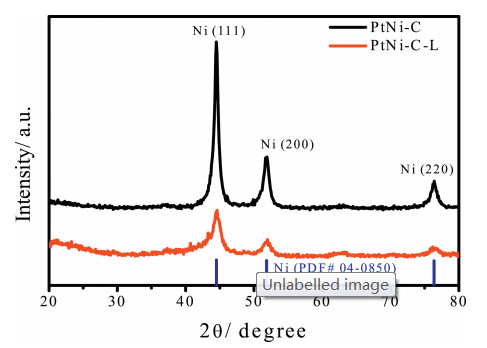
Figure 1.
Powder X-ray diffraction patterns for PtNi-C and PtNi-C-L catalysts.
Unstable Ni leaching in MOF-derived PtNi-C catalyst with improved performance for alcohols fuel electro-oxidation
Bo Fang , Zong Liu , Yufei Bao , Ligang Feng

Direct alcohol fuel cells (DAFCs) are promising energy supply devices in converting chemical energy into electric energy ecofriendly with high efficiency and energy density [1, 2]. Among the candidate fuels, methanol has been the most largely studied because it is more efficiently oxidized than other alcohols; ethanol that can be produced in large quantities from biomass is also an attractive alternative fuel. To realize the energy efficiently conversion, two half-reactions are fulfilled that includes the anode alcohol fuel oxidation and the cathode oxygen reduction reaction [3, 4]; compared with the cathode oxygen reduction, the anode alcohol fuel oxidation is more sluggish and high overpotentials required for alcohols oxidation results in low power generation. Attention should be direct to the anode side alcohol fuel oxidation, especially to realize its complete oxidation to CO2 and water.
Pt represents an important class of catalysts in terms of their unique catalytic activity in fuel cell technology for the electrooxidation of methanol, ethanol, glucose, oxygen reduction reaction, etc. [5, 6]. However, the pure Pt will inevitably suffer from the poisoning problems during the alcohol fuel oxidation process resulting in poor activity and stability [7, 8]. Considering its unique catalytic ability but limited resource on earth, it is urgent to increasing Pt catalytic efficiency for application in the fuel cells technique. Pt hybrid with other transition cheap metals in the form of alloy/doping/surface modification was considered very promising for the oxidation of alcohol fuels, because of the greatly increased catalytic performance and anti-poisoning ability. The catalytic performance of Pt alloy can be efficiently increased by means of engineering the alloy composition, structure size and morphology [5]. The increased catalytic performance is attributed to the improved oxophilic properties generated by the second component in the catalysts system, that will reduce the COintermediates poisoning problem.
Recently, metal - organic frameworks (MOFs) have attracted enormous attention in the catalyst fabrication, and the straightforward route is the simple carbonization to obtain the functional metal species that generally possess an extremely high surface area and heteroatom doping effect [9-11]. Furthermore, by varying the metal ions/clusters and organic linkers, different functional MOFs can be easily obtained that are very promising for the functionalized catalysts fabrication. For example, Pt - Ni bimetallic nanoparticles supported on MOF-5-derived carbon were reported very active as ORR electrocatalyst, and the high performance was attributed to Pt electron interaction with Ni and carbon support [12]. Hierarchical porous carbon nanofibers derived from ZIF-67 embedded into electrospun polyacrylonitrile were used as supports to anchor octahedral Pt-Cu nanocrystals and the as-prepared nanocomposites exhibited outstanding electrocatalytic performance in an acidic medium for hydrogen evolution [13]. Porous carbon prepared by direct carbonization of the porous metalorganic framework was used to support metallic PtM (M = Fe, Ni, Co, Cu) nanoparticles and a superior power density of 121 mW/cm2 at 90 ℃ was obtained on PtFe catalyst for direct ethanol fuel cells [14].
Inspired by the above work, PtNi catalyst comes into our mind because of its promising catalytic application in direct alcohol fuel cells. To our survey, PtNi based catalyst derived from the MOF materials was rarely concerned for alcohol fuel cells oxidation. In light of its high oxophilic property tuning by the different oxidation state of Ni in the system, herein, we carefully studied the PtNi hybrid catalyst for methanol and ethanol oxidation derived from the simple MOF materials by combining Pt nanoparticles with NiBTC after annealing in the tube furnace (named as PtNi-C). A relatively low catalytic performance for alcohol fuel oxidation was obtained on the pristine PtNi-C sample, while it was found the catalytic ability can be greatly enhanced by leaching the unstable Ni species in the hybrid catalysts (named as PtNi-C-L). Specifically, the peak current density was increased by 5.1 and 3.7 times for methanol and ethanol oxidation; the high catalytic performance can be attributed to the more Pt active sites exposure and maximized synergistic effect of Pt and Ni based on the bifunctional catalytic mechanism. The current work guides the importance of unstable assistant promoter removal for the MOF derived catalysts.
The details of the catalyst materials fabrication can be found in Supporting information. Briefly, the PtNi-C sample was prepared by combining Pt nanoparticles with Ni-MOF and the subsequent thermal annealing at 450 ℃ in N2 atmosphere [15]. To removal the unstable Ni species that was not coordinated with Pt, the pristine PtNi-C was soaked in 1 mol/L nitric acid for 2 h; and the recollected catalyst powder (PtNi-C-L) was comparatively studied with the pristine samples. The bulk composition of the powder catalysts was probed by inductively coupled plasma optical emission spectrometry (ICP-OES), and it was calculated that the absolute amount of Pt was almost not changed while the content of Ni was reduced for a given amount of PtNi-C catalyst due to the loss of unstable Ni species in the system. While the relative content of Pt was calculated to be 1.7 wt% and 3.0 wt% for PtNi-C and PtNi-C-L respectively. Because of the low amount of Pt in the system, the diffraction peaks assigned to Ni were mainly observed in the X-ray diffraction (XRD) patterns and no obvious peaks from Pt were responded in Fig. 1. The major diffraction peaks at the 2θ values around 44.5°, 51.8° and 76.4° were identified as Ni (111), (200) and (220) planes (PDF No. 04-0850). After unstable Ni leaching, the diffraction peak intensity of Ni was reduced and the diffraction peaks became wider compared with the pristine PtNi-C catalyst. The change in the XRD pattern indicated that the unstable Ni species in the large particles with high crystallinity were removed by acid leaching easily, and the rest Ni species in a more stable state were probably confined in the carbon structure and in the form of PtNi alloy.
The morphology of the sample was probed by transmission electron microscopy (TEM) technique, and nanoparticles were observed with the average particle size around 14 nm for the PtNiC sample (Fig. 2a and Fig. S1 in Supporting information). The lattice distance of 0.223 and 0.200 nm can be found assigning to PtNi (111) and Ni (111) plane (Fig. 2b) [16, 17]. A polycrystal structure of Ni was indicated in the selected area electron diffraction (SADE) (inset in Fig. 2c). The elemental mapping demonstrated the presence of C, Ni and Pt (Figs. 2e-g). While for the PtNi-C-L sample, most of the particles were leached out and a hollow shell-like structure was thus formed in Fig. 2a'. Some stable nanoparticles were still visible inside the carbon matrix encapsulated by the continuous graphite layer. This kind of structure was often observed in acid-treated MOF derived carbon [18, 19]. The lattice distance of 0.223, 0.200 and 0.330 nm can still be found for PtNi (111), Ni (111) and C (003) plane (Fig. 2b'), and the inset SADE pattern still indicated the polycrystalline structure of Ni (Fig. 2c'). The elements of C, Ni and Pt were still visible after the acid leaching (Figs. 2e'-g').

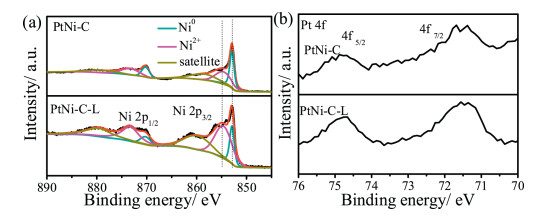
The surface chemical state of the samples was comparatively probed by X-ray photoelectron spectroscopy (XPS), and no obvious peak position change was found probably due to the low amount Pt in the catalyst system. The high resolution Ni 2p XPS spectrum consisted of two bands, namely 2p1/2 and 2p3/2, respectively (Fig. 3a); and each band can be deconvoluted into metallic Ni (852.92 eV) and Ni2+ (854.62 eV) as well as the satellite peak [20, 21]. Due to the low amount of Pt, the signal of Pt 4f was weakly responded; but the presence of Pt was clearly indicated by the Pt 4f spectra [22] (Fig. 3b). Because of the electronegativity differences between Ni and Pt, electron transfer from Ni to Pt was generally reported for the bulk PtNi alloy [23], Such an electron transfer can reduce the density of states on the Fermi level and thus improve the performance of PtNi alloys toward alcohol fuel oxidation by reducing the Pt-CO bond energy [23, 24]. The other point is that the oxophilic property of Ni could provide some oxygen-containing species in the form of nickel hydroxide/oxide during the methanol oxidation driven by high potential, that will contribute to the high performance according to the bi-functional catalytic mechanism for alcohols fuel and CO oxidation [23, 24]. Namely, during the catalytic process, the Pt services as the active sites for the alcohol molecular adsorption/dissociation, and the assistant component of oxophilic Ni offers the oxygen-containing species easily at lower potentials to help the reaction intermediates oxidation and removal.
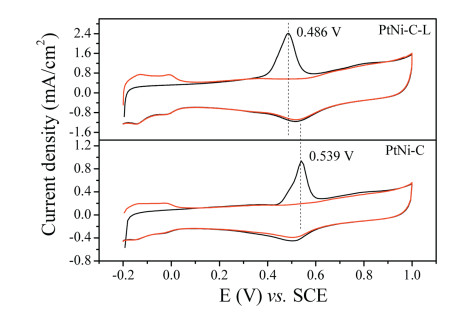
The high anti-CO poisoning ability was demonstrated by CO stripping voltammetry technique in Fig. 4. The onset potential and the peak potential for CO oxidation were 0.443 V and 0.539 V respectively for PtNi-C catalyst, and they were 0.357 V and 0.486 V for PtNi-C-L catalyst. After acid leaching, all the correspond potentials were shifted negatively. It was evident the pristine PtNiC catalyst was more likely to be poisoned. The exposed active sites could be estimated by the electrochemically active surface area (ECSA) by assuming 420 μC/cm2 charge required for a CO monolayer oxidation. It was calculated to be 59.77 m2/gPt and 101.9 m2/gPt for PtNi-C and PtNi-C-L respectively. The larger the ECSA value, the more the exposure of the active site. Due to the unstable Ni leaching, more Pt sites were exposed. The huge difference could be due to the composition change in the catalyst system resulting from the unstable Ni leaching. Because of the stable PtNi alloy trapped in the carbon layer, the excellent antipoisoning ability was thus obtained.
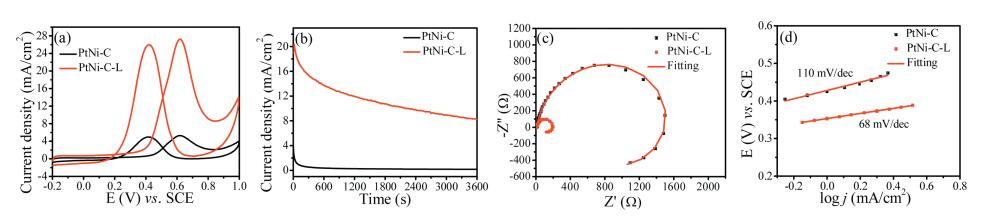
The methanol oxidation performance was studied by cyclic voltammetry in 0.5 mol/L H2SO4/1 mol/L CH3OH at the scan rate of 50 mV/s. As shown in Fig. 5a, the peak current density of the forward scan for PtNi-C-L was 27.28 mA/cm2, about 5.1 times higher than that of the PtNi-C electrode (5.3 mA/cm2). The onset potential for PtNi-C-L was 0.3 V, about 130 mV less than that of PtNi-C (0.43 V) indicating the highly improved ability for methanol fuel oxidation. As shown in Fig. S2a (Supporting information), the PtNi-C-L electrode showed higher specific activity (1.25 mA/cm2) than PtNi-C (0.74 mA/cm2). The PtNi-C-L electrode also exhibited higher mass activity (1273 mA/mgPt) than PtNi-C (440 mA/mgPt). The performance of PtNi-C-L was also much higher than other catalysts such as PtCo NCs (514.5 mA/mgPt) [25], PtNi/FCNTs (841.3 mA/mgPt) [26] and PtAuCu nanowires (928.1 mA/mgPt) [27]. The catalytic stability revealed by chronoamperometry (CA) at 0.6 V was shown in Fig. 5b. After 3600 s test, the final current density was 16.27 mA/cm2 for PtNi-C-L and 0.17 mA/cm2 for the PtNi-C catalyst, respectively. The decay rate by normalizing the lost current density to the initial value was 49.35%, much lower than that of 73.86% for PtNi-C catalyst. The electrochemical impedance was done to evaluate the charge transfer ability. At 0.4 V, a pseudoinductive behavior was indicated by a positive loop at higher frequencies following by a low frequency loop in the fourth quadrant (Fig. 5c). This is generally observed for methanol oxidation on Pt-catalyst because of the first dehydrogenated to form adsorbed CO species [28]. The smaller charge-transfer resistance was found on the PtNi-C-L catalyst as an indication of high intrinsic activity for methanol oxidation. This inductive behavior can be understood that the COads (adsorbed CO) coverage will be decreased by increasing the potential while some time is required to reach a new steady-state coverage after a potential perturbation [29]. By fitting the Nyquist plot using a typical equivalent circuit (Fig. S3 in Supporting information), the chargetransfer resistance of PtNi-C-L was calculated to be 574.2 Ω, smaller than that of PtNi-C (Table S1 in Supporting information). The Tafel slope of PtNi-C-L and PtNi-C for methanol oxidation was found to be 68 mV/dec and 110 mV/dec, respectively (Fig. 5d). Tafel slope of about 120 mV/dec means that the unit reaction involving the first electron transfer is the rate-determining step for methanol electrooxidation reaction, which can be considered as a splitting of the first C–H bond of CH3OH molecule with the first electron transfer [30, 31]. The Tafel slope of 60 mV/dec indicates the water activation step as the rate-determining step [32]. The smaller Tafel slope is an indication of improved catalytic kinetics. The methanol oxidation mechanism is generally described as below, and the species (M) represents as an active center for adsorption of OHads at lower potentials [11].
|
|
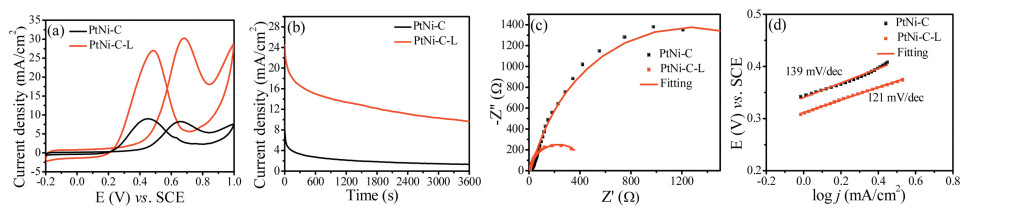
The ethanol oxidation performance was also studied by cyclic voltammetry in 0.5 mol/L H2SO4/1 mol/L C2H5OH at the scan rate of 50 mV/s. As shown in Fig. 6a, the peak current density of the forward scan for PtNi-C-L was 30.26 mA/cm2, about 3.7 times higher than that of the PtNi-C electrode (8.25 mA/cm2). The onset potential for PtNi-C-L was 0.31 V, about 50 mV less than that of PtNi-C (0.36 V) indicating the highly improved ability for ethanol fuel oxidation. As shown in Fig. S2b (Supporting information), the PtNi-C-L electrode showed much higher specific activity (1.39 mA/cm2) than PtNi-C (1.15 mA/cm2), and also the higher mass activity (1413 mA/mgPt) than PtNi-C (686 mA/mgPt). It was also better than some catalysts reported elsewhere, such as PtCu nanostars (629 mA/mgPt) [33], Pt/C-Cu3P (413.96 mA/mgPt) [34], Pt-CoSn/C (454.6 mA/mgPt) [35]. The catalytic stability revealed by chronoamperometry (CA) at 0.6 V was shown in Fig. 6b. The final current density was 9.66 mA/cm2 for PtNi-C-L and 1.31 mA/cm2 for the PtNi-C catalyst, respectively, after the 3600 s test. The decay rate by normalizing the lost current density to the initial value for PtNi-C-L was 44.51%, much lower than that of 64.31% for PtNi-C catalyst. The charge transfer resistance indicated by the diameter of the semicircle in the medium-frequency region for PtNi-C-L was also much smaller than that of PtNi-C as an indication of the rapid kinetics (Fig. 6c and Table S2 in Supporting information). The Tafelslope of PtNi-C-L and PtNi-C for ethanol oxidation was 121 mV/dec and 139 mV/dec, respectively (Fig. 6d). The smaller Tafel slope is an indication of improved catalytic kinetics, and the value of 120 mV/dec signified the first charge transfer for splitting of the first C–H bond as the rate-determining step [36, 37]. The above study demonstrated the significant catalytic performance improvement for the MOF-derived catalysts via easily acid leaching to remove the unstable species; this importance was not yet realized for the relevant catalyst fabrication and performance study.
In summary, the significance of unstable species leaching was demonstrated on MOF-derived catalysts, that was instructive for the relevant catalyst fabrication and performance study. For the proposed PtNi-C catalyst derived by combining Pt nanoparticles with Ni-BTC after annealing in the tube furnace, the unstable Ni species can be easily leached out in nitric acid for 2 h, and the stable PtNi nanoparticles trapped in the graphite carbon layer were obtained. The high electrochemically active surface area was found because of the Ni leaching induced porous structure. The improved catalytic ability for alcohol fuels oxidation was verified in terms of the high peak current density, specific and mass activity and rapid charge transfer kinetics and high catalytic stability.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The work is supported by the National Natural Science Foundation of China (Nos. 21603041, 21972124), the Priority Academic Program Development of Jiangsu Higher Education Institution. Z. Liu is thankful for the support of the Foundation of Excellent Doctoral Dissertation of Yangzhou University.
Supplementary material related tothisarticle canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.02.045.
X.P. Liu, Y.F. Zhang, S.F. Deng, et al., Chin. Chem. Lett. 30(2019) 299-304. doi: 10.1016/j.cclet.2018.09.021
H.M. Zhang, J. He, C.Y. Zhai, M.S. Zhu, Chin. Chem. Lett. 30(2019) 2338-2342. doi: 10.1016/j.cclet.2019.07.021
L.B. Hu, F. Yu, H.F. Yuan, et al., Chin. Chem. Lett. 30(2019) 624-629. doi: 10.1016/j.cclet.2018.10.039
F.L. Wang, H.G. Yu, Z.Q. Tian, H.G. Xue, L.G. Feng, J. EnergyChem. 27(2018) 395-403.
C.X. Xu, J.G. Hou, X.H. Pang, et al., Int. J. Hydrogen Energy 37(2012) 10489-10498. doi: 10.1016/j.ijhydene.2012.04.041
B.Y. Xia, H.B. Wu, X. Wang, X.W. Lou, J. Am. Chem. Soc.134(2012) 13934-13937. doi: 10.1021/ja3051662
E. Antolini, Energies 10(2017) 42. doi: 10.3390/en10010042
W.S. Chen, J. Xue, Y.F. Bao, L.G. Feng, Chem. Eng. J. 381(2020) 122752. doi: 10.1016/j.cej.2019.122752
B. Fang, L.G. Feng, Acta Phys.-Chim. Sin. 36(2020) 1905023. doi: 10.3866/PKU.WHXB201905023
W.H. Wang, Q. Gao, A.L. Li, et al., Chin. Chem. Lett. 29(2018) 336-338. doi: 10.1016/j.cclet.2017.08.033
Y. Yu, S.J. You, J.N. Du, et al., Appl. Cata. B:Environ. 259(2019) 118043. doi: 10.1016/j.apcatb.2019.118043
I.A. Khan, Y.H. Qian, A. Badshah, M.A. Nadeem, D. Zhao, ACS Appl. Mater. Interfaces 8(2016) 17268-17275. doi: 10.1021/acsami.6b04548
J.L. Zhang, W. Jia, S.Q. Dang, Y.L. Cao, J. Colloid Interface Sci. 560(2020) 161-168. doi: 10.1016/j.jcis.2019.10.062
I.A. Khan, A. Badshah, N. Haider, et al., J. Solid State Electrochem. 18(2014) 1545-1555. doi: 10.1007/s10008-013-2377-8
X.J. Xu, J. Liu, J.W. Liu, et al., Adv. Funct. Mater. 28(2018) 1707573. doi: 10.1002/adfm.201707573
C.M. Zhang, R.Z. Zhang, X.K. Li, W. Chen, ACS Appl. Mater. Interfaces 9(2017) 29623-29632. doi: 10.1021/acsami.7b04489
K. Zhang, Q.L. Yue, G.F. Chen, et al., J. Phys. Chem. C 115(2010) 379-389.
X.J. Wang, J.W. Zhou, H. Fu, et al., J. Mater. Chem. A 2(2014) 14064-14070. doi: 10.1039/C4TA01506A
S.H. Ahn, A. Manthiram, Small 13(2017) 1603437. doi: 10.1002/smll.201603437
Z. Liu, X. Yu, H.G. Yu, H.G. Xue, L.G. Feng, ChemSusChem 11(2018) 2703-2709. doi: 10.1002/cssc.201801250
C.L. Wang, C.Q. Song, W.H. Shen, et al., Catal. Sci. Technol. 9(2019) 1769-1773. doi: 10.1039/C9CY00191C
X.D. Yang, J. Xue, L.G. Feng, Chem. Commun. 55(2019) 11247-11250. doi: 10.1039/C9CC06004A
Y.J. Hu, P. Wu, Y.J. Yin, H. Zhang, C.X. Cai, Appl. Catal. B:Environ. 111-112(2012) 208-217.
F.L.Wang, B.Fang, X.Yu, L.G.Feng, ACSAppl.Mater.Interfaces11(2019)9496-9503. doi: 10.1021/acsami.8b18029
Q.L. Chen, Z.M. Cao, G.F. Du, et al., Nano Energy 39(2017) 582-589. doi: 10.1016/j.nanoen.2017.07.041
A.B.A.A. Nassr, I. Sinev, W. Grünert, M. Bron, Appl. Catal. B:Environ. 142-143(2013) 849-860.
Y.J. Liu, G.H. Ren, M.Q. Wang, et al., J. Alloys. Compd. 780(2019) 504-511. doi: 10.1016/j.jallcom.2018.12.016
J.F. Chang, L.G. Feng, C.P. Liu, W. Xing, X.L. Hu, Energy Environ. Sci. 7(2014) 1628-1632. doi: 10.1039/c4ee00100a
U. Krewer, M. Christov, T. Vidakovic', K. Sundmacher, J. Electroanal. Chem. 589(2006) 148-159. doi: 10.1016/j.jelechem.2006.01.027
X.L. Wang, H. Wang, R.F. Wang, Q.Z. Wang, Z.Q. Lei, J. Solid State Electrochem. 16(2011) 1049-1054.
G. Wu, L. Li, B.Q. Xu, Electrochim. Acta 50(2004) 1-10. doi: 10.1016/j.electacta.2004.07.006
S.L. Gojkovi c, T.R. Vidakovi c, Electrochim. Acta 47(2001) 633-642. doi: 10.1016/S0013-4686(01)00730-7
L.P. Huang, W. Zhang, P. Li, et al., Nano Res. 12(2019) 1147-1153. doi: 10.1007/s12274-019-2367-y
R.X. Li, Z.Z. Ma, F. Zhang, et al., Electrochim. Acta 220(2016) 193-204. doi: 10.1016/j.electacta.2016.10.105
H. Wang, X.T. Zhang, R.F. Wang, et al., J. Power Sources 196(2011) 8000-8003. doi: 10.1016/j.jpowsour.2011.05.062
A. Kowal, M. Li, M. Shao, et al., Nat. Mater. 8(2009) 325-330. doi: 10.1038/nmat2359
J. Willsau, J. Heitbaum, J. Electroanal, Chem. Interfacial Electrochem. 194(1985) 27-35. doi: 10.1016/0022-0728(85)87003-0

Figure 2 (a, a') TEM image, (b, b') high-resolution TEM, (c, c') SAED and (d, d') scanning TEM, (e-g, e'-g') elemental mapping images of the PtNi-C and PtNi-C-L catalysts, respectively.

Figure 4 CO stripping voltammograms of PtNi-C and PtNi-C-L catalysts in 0.5 mol/L H2SO4 solution at the scan rate of 20 mV/s.

Figure 5 (a) Cyclic voltammograms at 50 mV/s, (b) chronoamperometric curves at 0.6 V, (c) Nyquist plots at 0.4 V, (d) Tafel plots of PtNi-C and PtNi-C-L catalysts for methanol oxidation in 0.5 mol/L H2SO4 and 1 mol/L CH3OH, respectively.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

