Citation:
Zhu Tao, Yang Yongrui, Zhou Suyuan, Yao Xiang, Liu Lei, Hu Wenping, Gong Xiong. Bulk heterojunction perovskite solar cells incorporated with solution-processed TiOx nanoparticles as the electron acceptors[J]. Chinese Chemical Letters,
2020, 31(9): 2249-2253.
doi:
10.1016/j.cclet.2020.02.004
Bulk heterojunction perovskite solar cells incorporated with solution-processed TiOx nanoparticles as the electron acceptors
English
Bulk heterojunction perovskite solar cells incorporated with solution-processed TiOx nanoparticles as the electron acceptors
Department of Polymer Engineering, College of Polymer Science and Polymer Engineering, The University of Akron, Akron, OH 44325, United States
b.
Institute of Molecular Plus, Tianjin Key Laboratory of Molecular Optoelectronic Sciences, School of Science, Tianjin University and Collaborative Innovation Centre of Chemical Science and Engineering, Tianjin 300072, China
xgong@uakron.edu (X. Gong). 1 These authors contributed equally to this work.
Received Date:
29 December 2019 Accepted Date:
05 February 2020 Revised Date:
24 January 2020 Available Online:
15 September 2020
Abstract:
In the past ten years, perovskite solar cells were rapidly developed, but the intrinsic unbalanced charge carrier diffusion lengths within perovskite materials were not fully addressed by either a planar heterojunction or meso-superstructured perovskite solar cells. In this study, we report bulk heterojunction perovskite solar cells, where perovskite materials CH3NH3PbI3 is blended with solution-processed n-type TiOx nanoparticles as the photoactive layer. Studies indicate that one-step solution-processed CH3NH3PbI3:TiOx bulk-heterojunction thin film possesses enhanced and balanced charge carrier mobilities, superior film morphology with enlarged crystal sizes, and suppressed trapinduced charge recombination. Thus, bulk heterojunction perovskite solar cells by CH3NH3PbI3 mixed with 5 wt% of TiOx, which is processed by one-step method rather than typical two-step method, show a short-circuit current density of 20.93 mA/cm2, an open-circuit voltage of 0.90 V, a fill factor of 80% and with a corresponding power conversion efficiency of 14.91%, which is more than 30% enhancement as compared with that of perovskite solar cells with a planar heterojunction device structure. Moreover, bulk heterojunction perovskite solar cells possess enhanced device stability. All these results demonstrate that perovskite solar cells with a bulk heterojunction device structure are one of apparent approaches to boost device performance.
Since perovskite solar cells (PSCs) with a power conversion efficiency (PCE) of 3.8% were first reported by Miyasaka and his co-workers [1], PSCs have been drawn great attention in both academic and industrial sectors in the past ten years [2-5]. Over 25% PCE was reported by Korea Research Institute of Chemical Technology collaborated with Massachusetts Institute of Technology [6]. However, the intrinsic electronic properties, i.e., the diffusion length of the hole is greater than that of the electron (Leff, h+/Leff, e- >1), of hybrid perovskite, which restricted further boosting device performance of PSCs, was not fully addressed so far [7]. In order to balance charge carrier transporting, hightemperature sintered TiOx (or Al2O3) electron extraction layer (EEL) was used as the scaffold in the meso-superstructured PSCs, which was similar to that of dye sensitized solar cells [8-13]. On the other hand, fullerene or fullerene derivatives used as the EEL in PSCs with a planar heterojunction (PHJ) device structure to enhance the extraction of electrons to the cathode [14]. But the electrical conductivities of above EELs are still not good enough to enhance the separated electrons to be transported and then eventually be collected, thus PCEs of PSCs were still poor [15]. Towards the end, we reported PSCs with a bulk heterojunction (BHJ) device structure, where perovskite materials were mixed with n-type electron acceptors, for balancing charge carrier transport properties, and then boosting PCEs of PSCs [15, 16].
Based on our previous report on BHJ PSCs [15-18], we report BHJ PSCs in this study, where perovskite materials CH3NH3PbI3 is mixed with solution-processed n-type TiOx nanoparticles rather than organic molecules. Solution-processed n-type TiOx nanoparticles are selected as the electron acceptors since it possesses high electron mobility and has a good solubility in the solvent which is used for dissolving CH3NH3PbI3. It is found that CH3NH3PbI3:TiOx BHJ thin film possesses enhanced and balanced charge carrier mobilities, superior film morphology with enlarged crystal sizes, and suppressed trap-induced charge recombination. Thus, BHJ PSCs exhibit more than 30% enhancement in PCEs as compared with that of PHJ PSCs. In addition, BHJ PSCs possess enhanced device stability.
Poly(3, 4-ethylene dioxy-thiophene):poly(styrene sulfonate) (PEDOT:PSS, SCA 388-20) was purchased from Heraeus. Lead iodide (PbI2, 99.99.5% metals basis) was purchased from Alfa Aesar. Phenyl-C61-butyric acid methyl ester (PC61BM, 99.5%) was purchased from Solenne BV. Dimethylformamide (DMF, anhydrous, 99.8%), dimethyl sulfoxide (DMSO, anhydrous, 99.9%), toluene (anhydrous, 99.8%), and chlorobenzene (anhydrous, 99.8%) were purchased from Sigma-Aldrich. All materials are used as received without any further processing. Methyl-ammonium iodide (MAI, CH3NH3I) was synthesized in our laboratory [19-22].
Perovskite precursor solutionwas prepared by dissolving PbI2 and MAI (molar ratio of 1:1) into DMF:DMSO (volume ratio of 4:1) with theconcentrationof1 mol/L.TiOxprecursor solution was prepared by diluting tetrabutyl titanate (TBT) with isopropyl alcohol solution (concentration of 3 vol%). PC61BM was dissolved in chlorobenzene with a concentration of 20 mg/mL.
The pre-cleaned indium tin oxide (ITO)/glass substrates were treated by UV-ozone plasma for about 20 min. Then, a ~ 40 nm PEDOT:PSS thin filmwas spin-cast on the top of the substrates with a spin speed of 3500 rpm for 30 s, followed with thermal annealing at 150 ℃ for 10 min. After PEDOT:PSS coated substrates are cooled down to room temperature, and then transferred into the glovebox with a nitrogen atmosphere. Afterward, either pristine CH3NH3PbI3 thin film or the CH3NH3PbI3:TiOx BHJ composite thin film was prepared by one-step method rather than typical two-step method, i.e., either pristine CH3NH3PbI3 thin fi 3NH3PbI3:TiOx BHJ composite thin film was deposited on the top of PEDOT:PSS layer by spin-coat with a spin speed of 4000 RPM for 30 s from corresponding CH3NH3PbI3 solution or CH3NH3PbI3 mixed with TiOx solution (with 5% and 10% by weight). After that, thin filmwere thermally annealed at 100 ℃ for 10 min and then cooled down to room temperature. Afterward, ~ 40 nm PC61BM thin film was spin-coated on the top of photoactive layer with a spin speed of 1500 rpm for 30 s. Finally, ~100 nm Al was thermally deposited on the topofPC61BM thin film in vacuum system with a base pressure of 10-6 Torr. The device area was measured to be 0.045 cm2.
The current density versus voltage (J–V) characteristics of PSCs was tested by a Keithley model 2400 source measure unit. The light source was a Newport Air Mass 1.5 Global (AM1.5 G) full spectrum simulator with a light intensity of 100 mW/cm2. The external quantum efficiency (EQE) spectrum was obtained on the solar cell quantum efficiency measurement system (QEX10). The scanning electron microscopy (SEM) images were got from a field emission scanning electron microscopy (Model JEOL-7401). Impedance spectroscopy was obtained on an HP 4194A impedance/gainphase analyzer. Time-resolved photocurrent (TPC) measurement was done by the home-made set-up in our laboratory [23-26].
Schemes 1a and b display PHJ PSCs with a device configuration of ITO/PEDOT:PSS/CH3NH3PbI3/PC61BM/Al and BHJ PSCs with a device structure of ITO/PEDOT:PSS/CH3NH3PbI3:TiOx/PC61BM/Al. It is clear that the only difference in PHJ PSCs and BHJ PSCs is the photoactive layer. In PHJ PSCs, the photoactive layer is pristine CH3NH3PbI3 thin film, whereas, in BHJ PSCs, the photoactive layer is CH3NH3PbI3:TiOx thin film. Scheme 1c displays the highest occupied molecular orbit (HOMO) energy levels and the lowest unoccupied molecular orbit (LUMO) energy levels of CH3NH3PbI3, TiOx and PC61BM, and the work functions of ITO, PEDOT:PSS and Al, respectively [5, 19]. According to the band alignment, the introduction of TiOx would decrease the energy barrier between CH3NH3PbI3 thin film and the PC61BM EEL, which would benefit for electronsto be transported, resulting in an enhanced short-circuit current (JSC) from BHJ PSCs as compared with that of PHJ PSCs. Thus, n-type TiOx acts as the electron acceptors in BHJ PSCs, which is similar to that of BHJ organic solar cells [27].
Scheme 1
Scheme 1.
Device structures of perovskite solar cells with (a) a planer heterojunction and (b) bulk heterojunction device structures. (c) The LUMO and HOMO energy levels of CH3NH3PbI3 (MAPbI3 TiOx PC61BM and the work functions of ITO, PEDOT:PSS and Al, respectively.
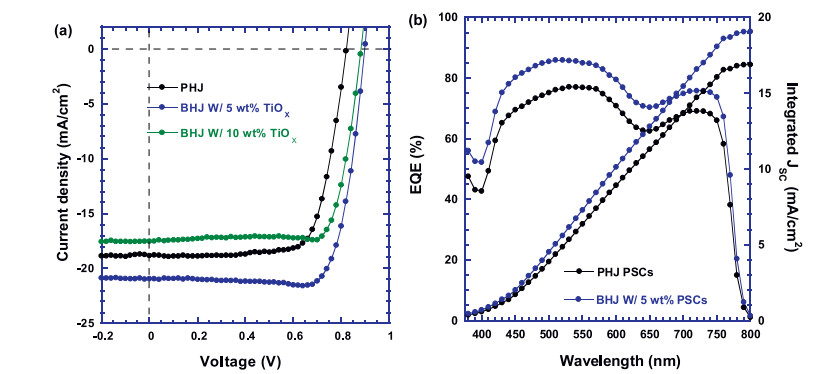
The J–V characteristics of PHJ and BHJ PSCs under white light illumination with a light intensity of 100 mA/cm2 are shown in Fig. 1a. The PHJ PSCs exhibit a JSC of 18.83 mA/cm2, an open-circuit voltage (VOC) of 0.82 V, a fill factor (FF) of 74% and with a corresponding PCE of 11.40%. All these device performance parameters are in good agreement with those reported values from PHJ PSCs, where CH3NH3PbI3 thin film was processed by onestep method [28]. The BHJ PSCs, where CH3NH3PbI3 mixed with 5 wt% of TiOx, show a JSC of 20.93 mA/cm2, a VOC of 0.90 V, a FF of 80% and with a corresponding PCE of 14.91%, which is more than 30% enhancement as compared with that of PHJ PSCs. However, as the ratio of TiOx is increased to 10 wt% in CH3NH3PbI3:TiOx thin film, the BHJ PSCs exhibit a JSC of 17.52 mA/cm2, a VOC of 0.88 V, a FF of 80% and with a corresponding PCE of 12.28%, which show poor device performance. Such poor device performance is probably attributed to the crystallinity of perovskites is tuned by the excess TiOx nanoparticles. The reproducibility of device performance is shown in Fig. S1 (Supporting information).
Figure 1
Figure 1.
(a) The J–V characteristics of PHJ PSCs by pristine CH3NH3PbI3 thin film and BHJ PSCs by CH3NH3PbI3 mixed with different ratios (5 wt% and 10 wt%) of TiOX thin films. (b) The EQE spectra of PHJ PSCs by pristine CH3NH3PbI3 thin film and BHJ PSCs by CH3NH3PbI3 mixed with 5 wt% of TiOX thin film.
The EQE spectra of both PHJ PSCs and BHJ PSCs are shown in Fig. 1b. It is obvious that BHJ PSCs possess larger EQE values than those of PHJ PSCs from 380 nm to 800 nm. Based on the EQE spectra, the integrated photocurrent densities are 16.90 mA/cm2 and 19.07 mA/cm2 for PHJ PSCs and BHJ PSCs, respectively. The photocurrent densities obtained from the EQE spectra are consistent with JSC values from the J–V characteristics (Fig. 1a). All these results demonstrate that TiOx as the electron acceptors could facility separated electrons to be efficiently transported, resulting in boosted device performance of BHJ PSCs.
In order to understand the underlying enhanced device performance of BHJ PSCs, SEM is carried out to study film morphologies of pristine CH3NH3PbI3 thin film and CH3NH3PbI3: TiOx BHJ composite thin film. Fig. 2 displays the top-view SEM images of pristine CH3NH3PbI3 thin film and CH3NH3PbI3:TiOx BHJ thin film. Lots of pinholes and grain boundaries are presented in pristine CH3NH3PbI3 thin film. As a result, PHJ PSCs exhibit poor device performance. On the contrast, a smoother and more compact surface is observed from CH3NH3PbI3:TiOx BHJ composite thin film, which indicates that the pinholes are probably filled with TiOx nanoparticles. Furthermore, the grain size of CH3NH3PbI3: TiOx BHJ composite thin film is ~ 300 nm, which is significantly larger than that (~100 nm) of pristine CH3NH3PbI3 thin film, reflecting that the introduction of TiOx nanoparticles could tune the crystallization rate and promote the growth of larger perovskite crystals. Superior film morphology and large grain size would facility charge carriers to be transferred in CH3NH3PbI3:TiOx BHJ composite thin film. Therefore, as expected, BHJ PSCs exhibit enhanced device performance.
Figure 2
Figure 2.
Top-view SEM images of (a) pristine CH3NH3PbI3 thin film and (b) CH3NH3PbI3:TiOx BHJ composite thin film.
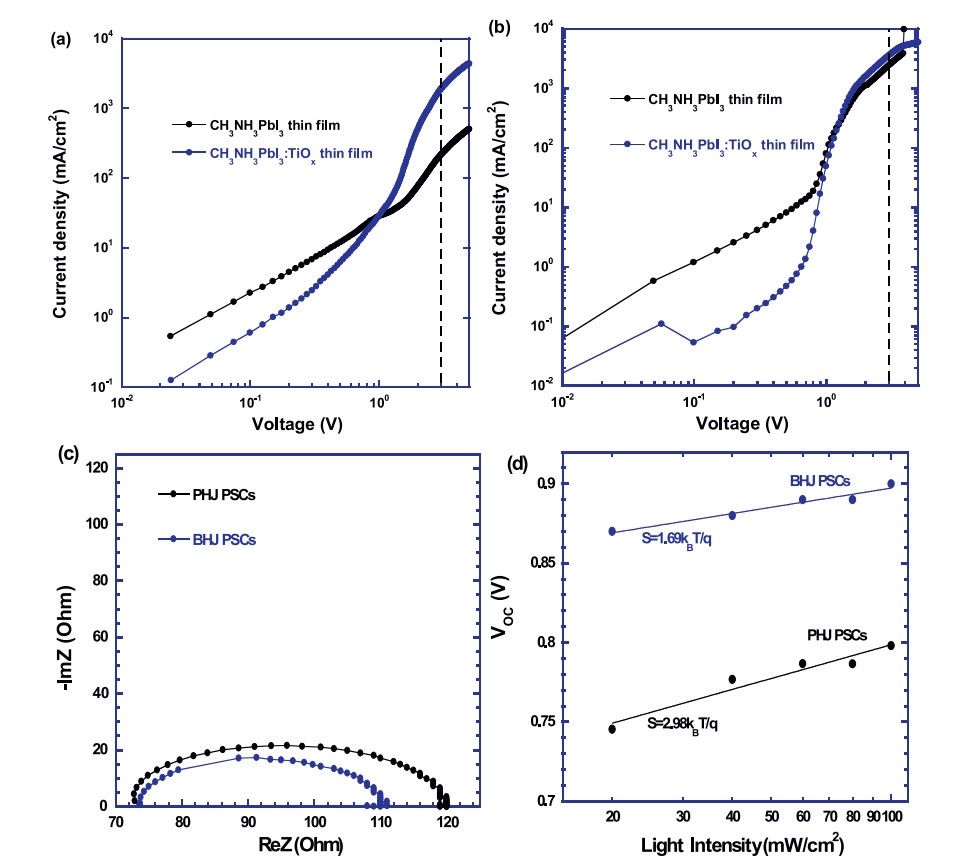
In order to investigate charge carrier transporting properties, charge carrier mobilities of pristine CH3NH3PbI3 thin film and CH3NH3PbI3:TiOx BHJ composite thin film are studied based on space charge limited current (SCLC) method according to MottGurney law [29-31]. The J–V characteristics of the electron-only diodes with a device structure of ITO/Al/active layer/PC61BM/Al and the hole-only diodes with a device structure of ITO/PEDOT: PSS/active layer/MoOX/Ag, where active layer is either pristine CH3NH3PbI3 thin film or CH3NH3PbI3:TiOx BHJ composite thin film, are shown in Figs. 3a and b. At a low voltage, the J–V curves are in the ohmic region, which indicates that the current is followed the Ohm's law. In the trap-filling region, the SCLC is dominated and which is described by [31], where J is the current density, e0 is the permittivity of free space (8.85 × 10-12 CV-1 m-1), ε is the relative dielectric constant of active material, μ is the mobility of charge carrier, V is the applied voltage and L is the thickness of active layer. In our study, the value of ε is 32 for CH3NH3PbI3 [20, 32] and the film thickness of CH3NH3PbI3 is ~ 300 nm, which was measured by DektakXT surface profile meter. For pristine CH3NH3PbI3 thin film, the hole mobility is measured to be 2.19 × 10-3 cm2 V-1 s-1), which is ten times higher than the electron mobility of 2.03 × 10-4 cm2 V-1 s-1. The charge carrier mobilities of pristine CH3NH3PbI3 thin film are consistent with reported values [32, 33]. The hole mobility larger than the electron mobility indicates that charge carrier transporting within pristine CH3NH3PbI3 thin film was unbalanced. For CH3NH3PbI3:TiOx BHJ composite thin film, the hole mobility is measured to be 3.47 × 10-3 cm2 V-1 s-1, which is nearly the same as the electron mobility (1.84 × 10-3 cm2 V-1 s-1, which is about ten times larger than that (2.03 × 10-4 cm2 V-1 s-1) of pristine CH3NH3PbI3 thin film. These results not only demonstrate that the electron mobility of CH3NH3PbI3 thin film is tuned by the TiOx electron acceptors, but also indicate that charge carrier transporting within CH3NH3PbI3:TiOx BHJ composite thin film is more balanced than those of pristine CH3NH3PbI3 thin film. As a result, BHJ PSCs by CH3NH3PbI3:TiOx BHJ composite thin film exhibit boosted JSC.
Figure 3
Figure 3.
The "log-log" plot of the J-V characteristics of (a) the electron-only diodes and (b) the hole-only diodes made by either pristine CH3NH3PbI3thin film or CH3NH3PbI3: TiOx BHJ composites thin film. (c) The Nyquist plots of PHJ and BHJ perovskite solar cells measured under a one-sun illumination at an applied voltage of VOC. (d) The light intensity dependence of VOC for PHJ PSCs and BHJ PSCs.
To further understand the underlying of enlarged JSC from BHJ PSCs, impedance spectroscopy (IS) is applied to investigate the electric properties of both PHJ PSCs and BHJ PSCs. In the IS spectra, the series resistance (RS) of PSCs is the sum of the electrodes' sheet resistance (Rsheet) and the internal charge transfer resistance (RCT) [18, 25, 34, 35]. Nyquist plots of both PHJ and BHJ PSCs under a onesun illumination at the voltage of VOC are shown in Fig. 3c. The RCT of PHJ PSCs is ~46 Ω, whereas, the RCT of BHJ PSCs is ~ 36 Ω. The smaller RCT, the larger JSC [18]. Thus, BHJ PSCs exhibit a larger JSC than that of PHJ PSCs.
The light intensity dependence of VOC is further studied for investigation of charge carrier recombination in both PHJ PSCs and BHJ PSCs. As shown in Fig. 3d, both PHJ PSCs and BHJ PSCs follow the relationship VOC∝/Sln(I) [36, 37], where S is the slope and I is the light intensity. For PHJ PSCs, S = 2.98kBT/q, where kB is the Boltzmann constant, q is the elementary charge and T is the room temperature in Kelvin, whereas, for BHJ PSCs, S = 1.69kBT/q. It was reported that a smaller S reflects that less trap-assisted charge recombination happens in PSCs. Thus, BHJ PSCs possess less trapassisted charge recombination, consequently higher JSC compared to that of PHJ PSCs.
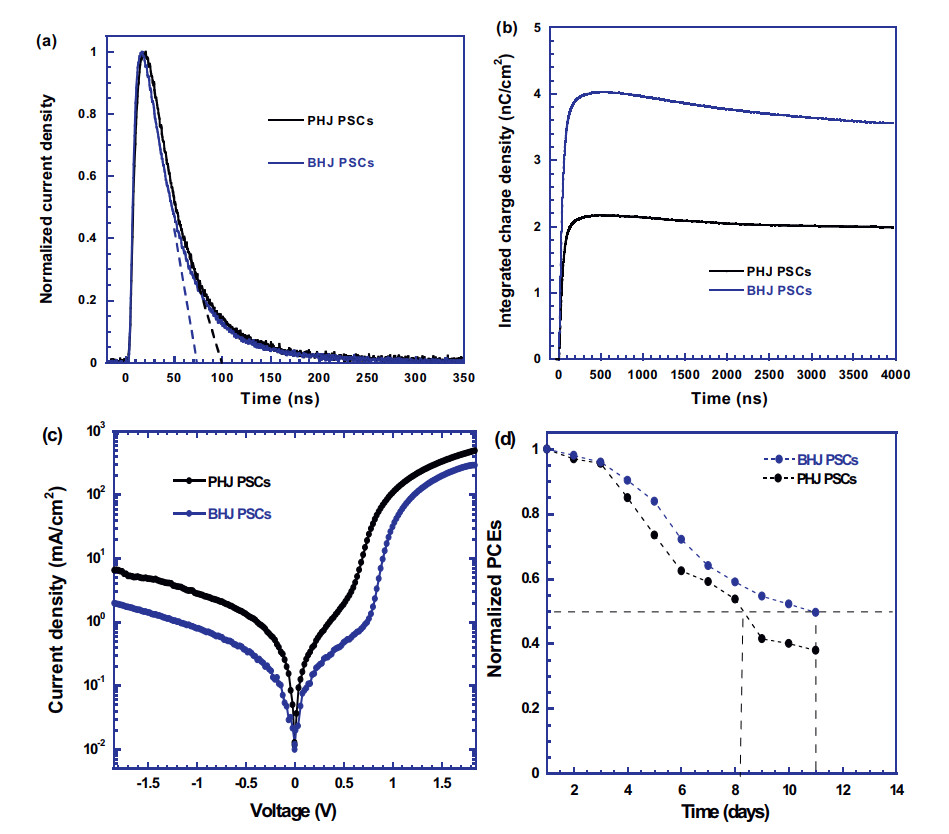
The transient photocurrent (TPC) measurement is further carried out to investigate the difference in charge extraction times between PHJ PSCs and BHJ PSCs. Fig. 4a shows the normalized TPC curves of PHJ PSCs and BHJ PSCs under -1 V bias, in which the charge carrier recombination process would be suppressed, and all the photocurrentis related to the charge extractionprocess. The charge extraction times for PHJ PSCs and BHJ PSCs are ~ 100 ns and ~ 75 ns, respectively. A shorter charge extraction time indicates that separated charge carriers are efficiently collected by the electrodes inBHJPSCs.The integrated charge densities versus times for bothPHJ PSCs and BHJ PSCs are shown in Fig. 4b. It is obvious that the total extraction charge densities of BHJPSCs are much larger than thoseof PHJ PSCs, indicating that more charge carriers are generated by BHJ PSCs rather than PHJ PSCs. All these results demonstrate that BHJ PSCs possess higher JSC as compared with that of PHJ PSCs.
Figure 4
Figure 4.
(a) The normalized TPC curves and (b) the integrated charge density curves of PHJ PSCs and BHJ PSCs, under bias of -1 V. (c) The J-V characteristics of both PHJ PSCs and BHJ PSCs measured in dark. (d) The self-stabilities of PHJ PSCs and BHJ PSCs.
To understand larger VOC observed from BHJ PSCs, the J–V characteristics of both PHJ and BHJ PSCs measured in dark are conducted and the results are shown in Fig. 4c. It is clear that BHJ PSCs possess lower dark current densities than those of PHJ PSCs under reverse biases. Based on the electronic properties of twoterm diodes, the VOC is described as [31, 38], where q is the elementary electron charge, kB is the Boltzmann's constant, T is the temperature, n is the ideality factor, J0 is the reverse dark current density, Jph is the photocurrent, respectively. Thus, lower dark current densities observed from BHJ PSCs indicate that BHJ PSCs exhibit larger VOC compared to that of PHJ PSCs.
In addition, the self-stabilities of PHJ PSCs and BHJ PSCs are primarily studied. Fig. 4d presents normalized PCEs of both PHJ PSCs and BHJ PSCs versus the time. It is found that the trends of decay in PCEs for both PHJ PSCs and BHJ PSCs are almost the same. However, BHJ PSCs still maintain about 50% PCEs of its original value after nearly 12 days while PHJ PSCs remain less than 50% PCEs of its original value only 8 days. These primary results indicate that BHJ PSCs possess good stability compared to that of PHJ PSCs. Such elongated stability is partially due to the superior film morphology of CH3NH3PbI3:TiOx BHJ composite thin film.
In conclusion, we reported high-performance bulk heterojunction perovskite solar cells, where CH3NH3PbI3 is blended with solution-processed n-type TiOx nanoparticle as the photoactive layer to balance charge carrier transporting properties for addressing the intrinsic electronic properties of perovskite materials. We found that the CH3NH3PbI3:TiOx bulk heterojunction thin film possesses enhanced and balanced charge carrier mobilities, superior film morphology with enlarged crystal sizes. Moreover, bulk heterojunction perovskite solar cells possess suppressed trap-induced charge recombination as compared with that of planar heterojunction perovskite solar cells. Thus, bulk heterojunction perovskite solar cells by CH3NH3PbI3 mixed with 5 wt% of TiOx show superior photovoltaic performance and better stability over planar heterojunction perovskite solar cells. All these results demonstrate that perovskite solar cells with a bulk heterojunction device structure are one of the facile approaches to boost device performance.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors at The University of Akron acknowledge Air Force Scientific Research Program (No. FA9550-15-1-0292) and National Science Foundation (Nos. EECs 1351785 and EECs 1903303) for financial support.
G. Wetzelaer, M. Kuik, M. Lenes, P. Blom, Appl. Phys. Lett. 99(2011) 153506. doi: 10.1063/1.3651752
Scheme 1
Device structures of perovskite solar cells with (a) a planer heterojunction and (b) bulk heterojunction device structures. (c) The LUMO and HOMO energy levels of CH3NH3PbI3 (MAPbI3 TiOx PC61BM and the work functions of ITO, PEDOT:PSS and Al, respectively.
Figure 1
(a) The J–V characteristics of PHJ PSCs by pristine CH3NH3PbI3 thin film and BHJ PSCs by CH3NH3PbI3 mixed with different ratios (5 wt% and 10 wt%) of TiOX thin films. (b) The EQE spectra of PHJ PSCs by pristine CH3NH3PbI3 thin film and BHJ PSCs by CH3NH3PbI3 mixed with 5 wt% of TiOX thin film.
Figure 3
The "log-log" plot of the J-V characteristics of (a) the electron-only diodes and (b) the hole-only diodes made by either pristine CH3NH3PbI3thin film or CH3NH3PbI3: TiOx BHJ composites thin film. (c) The Nyquist plots of PHJ and BHJ perovskite solar cells measured under a one-sun illumination at an applied voltage of VOC. (d) The light intensity dependence of VOC for PHJ PSCs and BHJ PSCs.
Figure 4
(a) The normalized TPC curves and (b) the integrated charge density curves of PHJ PSCs and BHJ PSCs, under bias of -1 V. (c) The J-V characteristics of both PHJ PSCs and BHJ PSCs measured in dark. (d) The self-stabilities of PHJ PSCs and BHJ PSCs.


 DownLoad:
DownLoad:




 下载:
下载:






