Citation:
Zhao Xiaoyu, Zhao Huachao, Sun Jiefang, Li Gang, Liu Rui. Blocking the defect sites on ultrathin Pt nanowires with Rh atoms to optimize the reaction path toward alcohol fuel oxidation[J]. Chinese Chemical Letters,
2020, 31(7): 1782-1786.
doi:
10.1016/j.cclet.2020.01.005
Blocking the defect sites on ultrathin Pt nanowires with Rh atoms to optimize the reaction path toward alcohol fuel oxidation
English
Blocking the defect sites on ultrathin Pt nanowires with Rh atoms to optimize the reaction path toward alcohol fuel oxidation
Tianjin Key Laboratory of Brine Chemical Engineering and Resource Eco-utilization, College of Chemical Engineering and Materials Science, Tianjin University of Science and Technology, Tianjin 300457, China
b.
State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
c.
Beijing Key Laboratory of Diagnostic and Traceability Technologies for Food Poisoning, Beijing Center for Disease Prevention and Control, Beijing 100013, China
Received Date:
29 November 2019 Accepted Date:
03 January 2020 Revised Date:
23 December 2019 Available Online:
15 July 2020
Abstract:
Anodic electrocatalyst plays the core role in direct alcohol fuel cells (DAFCs), while traditional Pt-catalysts suffer from limited catalytic activity, high over potential and severe CO poisoning. Herein, by selectively depositing Rh atoms on the defective-sites of Pt nanowires (NWs), we developed a new Pt@Rh NW electrocatalyst that exhibited enhanced electrocatalytic performance for both methanol oxidation (MOR) and ethanol oxidation (EOR). Both cyclic voltammetry (CV) and in-situ infrared spectroscopy revealed that the presence of Rh atoms suppressed the generation of poisonous intermediates and completely oxidized alcohols molecule into CO2. Atomic resolusion spherical aberration corrected high-angle annular dark field scanning transmission electron microscopy (CS-HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) mapping analysis revealed that Rh atoms were primarily deposited on the defective sites of Pt NWs. Meanwhile, the presence of Rh atoms also modified the electronic state of Pt atoms and therefore lowered the onset potential for alcohols oxidation potential. This work gives the first clear clue on the role of the defective sites of Pt nanocatalyst poisoning, and propose that selectively blocking these sites with trace amount of Rh is an effective strategy in designing advantageous electrocatalysts.
Fuel cells have attracted worldwide attention due to their potential applications in transportation and portable electronics [1, 2]. In comparison to hydrogen fuel cells, direct alcohol fuel cells (DAFCs) combine multiple advantages such as easy to transport, safety in use, and high energy density. In addition, alcohol fuels can be derived from renewable sources (i.e., fermentation of biomass and photoreduction of CO2) as well as from syngas, which provide a much lower cost compared to that of hydrogen fuel cells [3-5]. Anodic electrocatalyst plays the core role in DAFCs. However, low activity and poor stability of the catalysts limit the practical application of the DAFCs [6, 7]. The reason is that it not only comprises a large portion of the total cost by using the platinum group metal nanomaterials, but suffers from severe poisoning during the electrocatalyzed oxidation of alcohol and other small organic molecules [8]. This process does not result in the direct release of the chemical energy of the reactants, which substantially affects the fuel utilization efficiency. Moreover, the side products often exhibit higher binding energy on the reactive sites over ethanol itself, resulting in gradual poisoning/deactivation of nanocatalyst [9, 10].
To solve these problems, various strategies have been proposed to improve the activity and durability of nanocatalysts [11-15]. The successful examples involved control the morphologies of the nanocrystal and exposing specific highly active facets [16]. Coating the catalytically active Pd or Pt atoms outside Au to form a coreshell nanostructure is also an effective strategy [17]. In addition, introducing the second metal forming an alloy nanostructure can provide another promising solution to improve activity/durability of the Pt monometallic nanocatalyst [18]. All of these researches push great progresses in improving the performance of the catalyst/electrocatalyst, with large margin increased mass activity. However, only a few studies reported the complete oxidation of small alcohols, which involved the use of large amount of costly platinum family elements [19-21] to regulate the electronic structures of Pt/Pd nanomaterials, thus providing an optimized adsorption and catalytic oxidation for small alcohols [22].
Previous study demonstrated that the specific sites of the noble metal catalysts, defective sites, usually exhibit different catalytic performances. For example, defects on Pd nanocrystals, such as a twin boundary and stack fault, displays excellent activity for stabilizing atomic hydrogen, which is approximately 20 times more active than Pd {111} facet in the electrocatalytic hydrodehalogenation reaction [23]. We hypothesized that the electrooxidation of alcohols catalyzed on Pt atoms in the defective sites may follow different reaction pathways with their counterparts on crystal facets. To testify whether this hypothesis holds, Rh atoms were selectively deposited on the defective sites of Pt NWs by a similar protocol with the synthesize of Pd@Rh NWs [23]. The Pt@Rh NWs were systematically characterized with atomic resolution spherical aberration corrected high-angle annular dark field scanning transmission electron microscopy (CS-HAADFSTEM) and energy-dispersive X-ray spectroscopy (EDS) mapping. The defective sites on Pt NWs were blocked via the selective deposition of different amounts of Rh atoms, thus the role of defects was determined in the production of poisoning byproducts. The reaction process exhibited a low initial potential, moreover surface poisoning of the catalyst was relieved. Furthermore, we employed in situ infrared spectroscopy to monitor the catalysis reaction. It was indicated that the Pt@Rh NWs could completely oxidize methanol and ethanol to produce CO2, which would substantially improve fuel utilization.
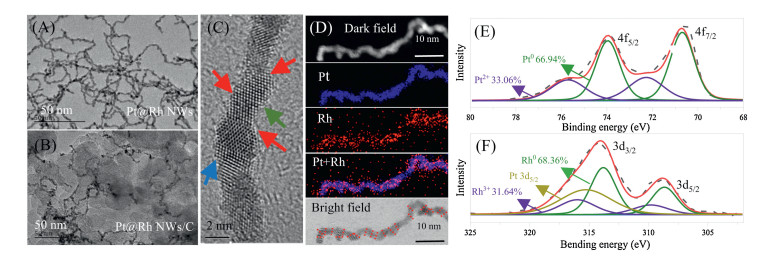
The Pt NWs were synthesized according to our previously reported procedure [23]. And then RhCl3 solution was added dropwise to 10.0 mL of freshly synthesized Pt NWs under stirring. The growth of Rh atoms was mainly achieved by chemical reduction. The KBH4 residue in the freshly synthesized Pt NWs dispersion was sufficient to reduce the gradually added metal salt RhCl3, and the resulting Rh atoms were preferential growth on the Pt NWs. TEM images show that ultrafine polycrystalline Pt and Pt@Rh NWs with rich defects were successfully obtained (Fig. 1A and Fig. S1 in Supporting information). With the increase of the Rh amount, the diameter of the synthesized nanowire catalysts also slightly increased accordingly. Specifically, the diameters of the Pt NWs, Pt@Rh 15%, and Pt@Rh 50% (Fig. S2 in Supporting information) were 2.43 ± 0.38 nm, 2.48 ± 0.409 nm and 2.77 ± 0.505 nm, respectively. This increase in the diameters provides a direct evidence of the Rh coating on the PtNWs. Of note is, when the deposited Rh was less than 15%, no distinct change was observed in morphology, partially due to the deposited Rh atoms being located at the defective sites. Furthermore, the Pt@Rh NW can be uniformly loaded onto conductive carbon black (Fig. 1B), very facile for their usage as electrocatalyst. Fig. 1C shows the high-resolution transmission electron microscopy (HRTEM) image of a single Pt@Rh NWs. Similar to the PtNW itself, dense defect sites, such as stacking faults (SF, blue arrows) and twin boundaries (TB, red arrows), and lattice defects (green arrows) are distributed on the NWs. These defects are formed by the directional oriented attachment of the Pt clusters during their liquid phase growth process. The X-ray diffraction (XRD) patterns for the Pt@Rh samples shown in Fig. S3 (Supporting information) are consistent with the standard Pt (JCPDS No. 04-0802) and Rh (JCPDS No. 05-0685).
Figure 1
Figure 1.
TEM of (A) Pt@Rh NWs and (B) Pt@Rh NWs/C; (C) HRTEM image of Pt NWs highlighted the presence of rich defect sites with arrows; (D) elemental mapping of Pt@Rh NWs using HAADF-STEM-EDS pattern; XPS spectra of Pt 4f (E) and Rh 3d (F).
To direct locating the Rh atoms, the Pt@Rh NWs were further observed by CS-HAADF-STEM. However, for the lower Z of Rh over that of Pt, we could not distinguish Rh atoms from Pt atoms based on their difference in Z contrast in HAADF-STEM image. Therefore, we manage to study the distribution of Rh atoms on Pt@Rh NWs by EDS mapping (Fig. 1D and Fig. S4 in Supporting information). By combining the EDS mapping and the bright-field image, where the defect sites were marked with red lines, it can conclude that Rh atoms is primarily deposited on the defective sites of the catalyst. Meanwhile, X-ray photoelectron spectroscopy (XPS) analysis revealed that the presence of Rh adatoms had neglectable influence on the electronic structure of Pt atoms, as neither the Pt 4f or Rh 3d spectrums of the Pt@Rh NWs changed upon the formation of core-shell structured NWs (Figs. 1E and F and Fig. S5 in Supporting information). Moreover, detailed curve fitting revealed that 70% of the Pt and Rh existed as Pt0 and Rh0 in the Pt@Rh NWs, which was in the same order with their monometallic counterparts. Therefore, the developed Pt@Rh is an ideal paradigm in studying the role of defective sites in the electronic process.
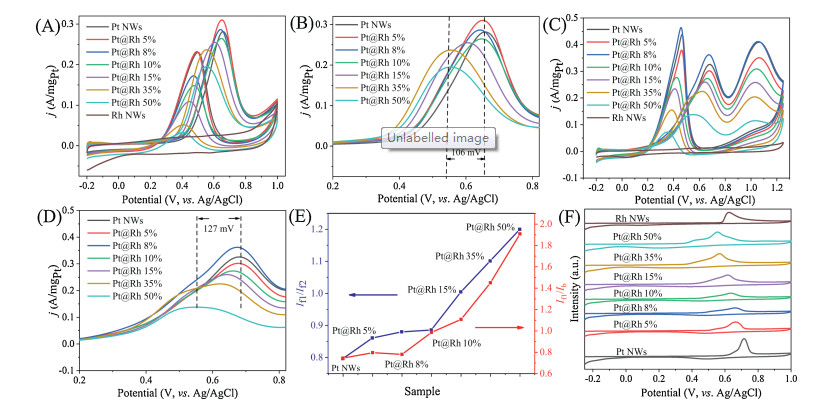
More specifically, the selectively blockage of Pt defective sites on NWs with Rh, the catalytic activity of the Pt@Rh NWs with different amounts of Rh in the electrooxidation of alcohols was studied. As shown in Fig. 2A, the Pt NWs are highly active in the methanol oxidation reaction (MOR), with the current density reached to 0.287 A/mgPt in the forward scan. However, the current density in backward scan (i.e., 0.232 A/mgPt) indicates the incomplete oxidation of methanol and the generation of a high concentration of CO and HCOOH [4, 24]. With the presence of trace amount of Rh atoms on Pt NWs, partial of Pt surface was blocked. Since Rh itself was completely inactive in this reaction, a slightly decrease in the activity of Pt@Rh NWs was observed. As reflected in the higher degree of decrease in the backward scan, the amounts of incomplete oxidation products were also decrease. However, the current peak density in the backward scan decreased substantially, caused by the surface blockage effect of Rh atoms. When the amount of Rh reached to 50%, the peak in the backward scan was almost completely suppressed, which indicated that the presence of surface Rh atoms effectively enhanced the reaction selectivity to CO2 [18, 25]. Therefore, we proposed that the degree of catalyst poisoning was effectively reduced by using Rh atoms to block the defect sites. In addition, the peak potential in the forward scan also decreased gradually with the increase of Rh amount, and for Pt coated with the highest amount of Rh, the potential of Pt@Rh 50% was 106 mV more negative than that of Pt NWs (Fig. 2B), which indicated the presence of Rh also improved the electrocatalytic performance.
Figure 2
Figure 2.
CVs of Pt@Rh NWs/C and Rh NWs/C catalysts for (A, B) MOR in 0.5 mol/L methanol and 0.1 mol/L HClO4 at 50 mV/s and (C, D) EOR in 0.5 mol/L ethanol and 0.1 mol/L HClO4 at 50 mV/s; (E) If1/If2 and If1/Ib values derived from EOR CVs; (F) CO-stripping curves of Pt@Rh NWs/C and Rh NWs/C catalysts.
Furthermore, the Pt@Rh NWs also exhibit high selectivity in the ethanol oxidation reaction (EOR) (Fig. 2C). Unlike the simple case for the MOR with only one electrooxidation peak, the electrooxidation of ethanol produces two peaks, the first peak (If1) is typically attributed to the formation of acetaldehyde, acetic acid and CO2, and the second peak (If2) is attributed to the formation of acetic acid [26]. The surface adsorbed incomplete oxidation products were oxidized in the reverse scan, and reflected in the backward scan current peak (Ib) [18]. Similarly with the case of MOR, the presence of Rh atoms lowered the current density in the forward scan but resulted in the downshift of the peak potential by approximately 127 mV (Fig. 2D). The relative intensity of If1 and Ib (If1/Ib) is extensively used as an indicator to assess the degree of incomplete oxidation carbonaceous intermediates accumulated on the catalyst surface, while the If1/If2 value evaluates the ability of the catalyst to cleave the C—C bond of ethanol [12]. Based on the Fig. 2E, the If1/Ib and If1/If2 values on the Pt@Rh NWs are significantly higher than that on the Pt NWs. The results indicate that the addition of Rh inhibits the poisoning of the Pt@Rh NWs/C catalyst surface and promotes cleavage of the C—C bonds. The improved anti-poisoning capacity of Pt@Rh NWs is further supported by the CO-stripping experiment (Fig. 2F and Fig. S6 in Supporting information). The CO oxidation peak on the Pt NWs catalyst is located at approximately 0.71 V, and the oxidation peak on Rh is located at 0.62 V. After Rh deposition on Pt, the CO oxidation peak is broadened and downshifted, indicating part of the strong binding sites were blocked. Moreover, the CO oxidation potential of the Pt@Rh 50% catalyst is 0.55 V, which is 160 mV lower than that of the Pt NWs and 90 mV lower than that of the Rh NWs. Therefore, the presence of Rh increased the anti-poisoning capacity of Pt NWs by a large margin.
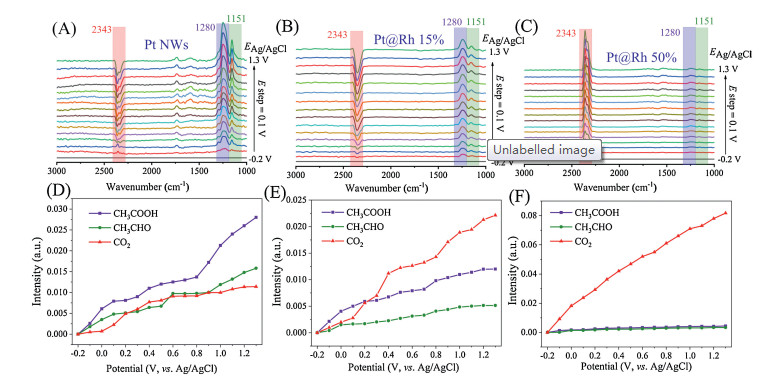
To gain a deeply understanding of the electrooxidation process, electrochemical in situ FTIR studies were performed on these Pt@Rh catalysts (Fig. 3 and Fig. S7 in Supporting information). Various absorption bands appear in the spectrum as the electrode potential increases. According to previous studies [6, 27], the band at 2343 cm-1 assigned to the asymmetric tensile vibration of CO2 is associated with complete oxidation of the alcohols [22]. The IR band at 1151 cm-1 is indicative of forming formaldehyde and acetaldehyde, and the other band at 1280 cm-1 belongs to the C—O tensile deformation indicating to the formation of carboxylic acid [28] (Table S1 in Supporting information). The right side of the figure shows the in situ infrared spectrum intensity statistics of different products. For the EOR on the Pt NWs catalyst (Figs. 3A and D), only a small amount of CO2 is produced, and majority of the products are acetaldehyde and acetic acid. When the amount of Rh increased to 15% (Figs. 3B and E), CO2 became the major products, and with the further increase of Rh amount to 50% (Figs. 3C and F), almost no acetaldehyde and acetic acid were formed in the reaction, and nearly all of the ethanol molecules were completely oxidized to CO2. Therefore, the selectivity of the Pt@Rh catalyst for CO2 and CH3COOH is very sensitive to the mount of Rh atoms. As the number of external Rh atoms increased, the selectivity toward CO2 gradually increased, and Pt@Rh 50% exhibited the highest CO2 selectivity. The surface of the Pt NWs catalyst contains abundant defect sites, and these sites possess excellent electrocatalytic activity. However, the Rh atoms take up the defect site and reduces the adsorption of the intermediate on the defects, which is a key factor for decreasing the poisoning effect. This result is consistent with the results obtained from the CO-stripping experiment and cyclic voltammetry (CV) curve, indicating that the Rh atom is beneficial for improving the complete oxidation ability of the catalyst. Therefore, the defect site on the Pt NWs is very active in the electrooxidation process of alcohol fuel, but also suspicious for poisoning. The Rh atom blocking strategy effectively regulates the defect richness, inhibits the formation of poisoning intermediates and promotes the complete oxidation of the alcohol fuel.
Figure 3
Figure 3.In situ FTIR spectra of the electrocatalysts for EOR and band intensities of CO2 (2343 cm-1), CH3COOH (1280 cm-1) and CH3CHO (1151 cm-1).
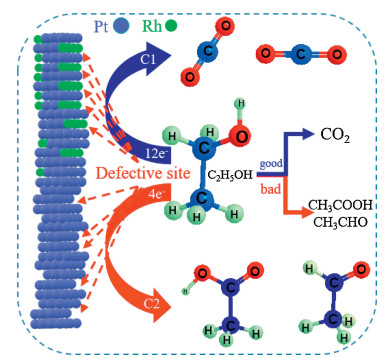
It is demonstrated that Pt@Rh NWs has excellent electrocatalytic performance in the process of electrooxidizing alcohol fuel. Based on the previously mentioned experimental data, we propose a reaction mechanism (Fig. 4). Due to the surface containing abundant defect sites with high adsorption energies, Pt NWs exhibit a strong dehydrogenation ability, which can rapidly oxidize alcohol fuel to form carboxylic acid and aldehyde molecules and the electrooxidation process primarily involves a C2 pathway. Therefore, the defect sites can effectively adsorb reactants for electrooxidation and act as the main active site in the reaction process. Meanwhile, the as-form intermediates, such as CO, adsorbed on the surface of the catalyst, resulting in poisoning of the catalyst. Interestingly, the selectively deposition of Rh atoms could significantly enhance the anti-poisoning capacity, by effectively regulated the structure of the defect sites. In addition, water molecules on the Pt surface are easily activated to form intermediate species (i.e., OHads) [29, 30]. Rh exhibits a strong oxygen absorption ability and can adsorb OHads in solution. These materials can further oxidize the intermediate products in the reaction process. Therefore, the alcohol fuel can be fully oxidized to produce CO2, and the energy of the reaction process can be fully converted. What is more, our strategy of sealing defect sites by the Rh atoms can effectively improve the reaction path, thus transforming the reaction path from C2 to C1 and substantially improves the CO2 selectivity.
Figure 4
Figure 4.
Proposed mechanism for EOR on Pt@Rh NWs electrocatalysts.
In summary, we demonstrated the successful preparation of defect-regulated Pt@Rh NWs, which use as effective oxidation electrocatalysts for alcohol fuel. It was verified that the defect sites were both acting as the reactive site and the main point of poisoning. By loading different amounts of Rh atoms onto the defect sites, the oxidation potential was effectively reduced, the accumulating of poison intermediates was also suppressed. What is more, the Rh atom effectively altered the structure of the defect site and improved the ability of the catalyst to electrooxidize the alcohol fuel. The catalyst not only completely oxidizes methanol but also promotes C—C bond cleavage of ethanol, resulting in their complete oxidation. Therefore, the defect blocking strategy significantly improved the activity of the catalyst as well as the product selectivity. In addition, the reaction process converts from the C2 path to the C1 path. This change allows the energy from the oxidation of the alcohol fuel to be completely converted. This work provides a new basis for the design and synthesis of novel catalysts for alcohol fuel oxidation.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was financially supported by the Natural Science Foundation of Tianjin Municipality (No. 18JCYBJC21200).
J.C. Dong, X.G. Zhang, V. Briega Martos, et al., Nat. Energy 4 (2019) 60-67. doi: 10.1038/s41560-018-0292-z
Figure 1
TEM of (A) Pt@Rh NWs and (B) Pt@Rh NWs/C; (C) HRTEM image of Pt NWs highlighted the presence of rich defect sites with arrows; (D) elemental mapping of Pt@Rh NWs using HAADF-STEM-EDS pattern; XPS spectra of Pt 4f (E) and Rh 3d (F).
Figure 2
CVs of Pt@Rh NWs/C and Rh NWs/C catalysts for (A, B) MOR in 0.5 mol/L methanol and 0.1 mol/L HClO4 at 50 mV/s and (C, D) EOR in 0.5 mol/L ethanol and 0.1 mol/L HClO4 at 50 mV/s; (E) If1/If2 and If1/Ib values derived from EOR CVs; (F) CO-stripping curves of Pt@Rh NWs/C and Rh NWs/C catalysts.

 DownLoad:
DownLoad:

 下载:
下载:





