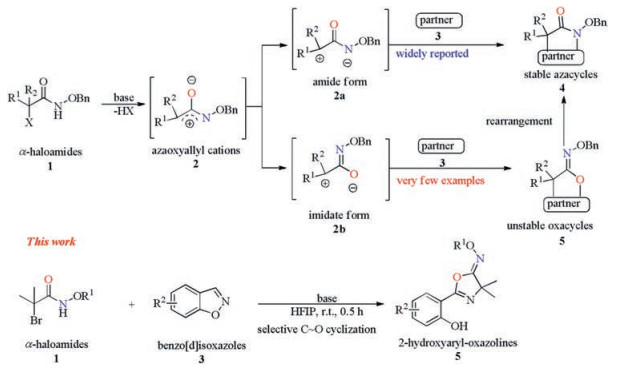
Scheme 1.
Multitasking annulation modes of azaoxyallylic cations.

1, 3-Dipolar cycloadditions represent a powerful tool in organic chemistry because efficient and atom-economical method for constructing various heterocycles or carbocycles could be provided [1]. Among which, the azaoxyallylic cation 2 [2] generated in situ from α-haloamide 1 by elimination of hydrogen halide has been recently drawn considerable attention [3]. Initially, Jeffrey reported the synthesis of seven-member N-heterocycles via [4 + 3] cycloaddition of the azaoxyallylic cations with furan [4]. Since then, the [3 + m] cycloaddition of the azaoxyallylic cations 2 with cyclization partners [5] has been widely developed as a wise method to construct N-heteroaromatic compounds 4, such as indole derivatives [6], carbonyl compounds [7] and sulfur ylides [8]. Theoretically, the azaoxyallylic cations 2 exist as two tautomers: amide form 2a and imidate form 2b. Chemists mainly focus on amide form 2a as C~N 3-atom synthon to afford azacycles 4. However, the study on imidate form 2b as C~O 3-atom synthon to construct oxa-heterocycles is less developed [5b, 5j, 6d, 7a, 7e] because the corresponding product iminolactone take easily rearrangement to give lactams 4 [9]. We assumed that, imidate form 2b of azaoxyallylic cations 2 could be obtained as a main tautomer under the suitable reaction conditions and react with suitable cyclization partners to give the oxacycles 5 (Scheme 1).
To verify this hypothesis, the reaction of 2-bromo-propana-mides 1 and benzo[d]isoxazoles 3 [10] was chosen as the reaction substrates to synthesize 2-hydroxyaryl-oxazolines 5, which is a privileged skeleton in a diverse array of biologically active molecules, such as antitumour, antibiotic and antitrypanosomally alkaloids [11]. The conventional approaches toward such a core suffer from multi-steps and harsh reaction conditions [12]. Thus, the development to efficiently build 2-hydroxyaryl-oxazolines from readily available starting materials in one pot manner under mild conditions remains significant challenge and highly desirable. Based on our research interest on construction of heterocycle [13], we explore herein a novel and effective base-mediated one-pot synthesis of hydroxyaryloxazolines 5 starting from α-haloamides 1 and benzo[d]isoxazoles 3 under mild and simple conditions via ring opening of benzo[d]isoxazoles, selective [3 + 2] cycloaddition and H-transfer cascade reaction. The significance of this protocol is not only due to its great potential for accessing a wide range of 2-hydroxyaryl-oxazolines but also the usage of the azaoxyallylic cations as the imidate 2b tautomer of azaoxyallylic cation to construct oxa-heterocycles. During preparation of this manuscript, Feng reported similar work [14]. Compared to Feng's work, this method is advantageous with weak base and short reaction time.
We initiated our investigation on the model reaction of N-(benzyloxy)-2-bromo-2-methylpropanamide (1a) and benzo[d]-isoxazole (3a) to optimize various reaction parameters. The results are summarized in Table 1. The designed ring opening annulation was carried out in the absence of transition metal as a catalyst at room temperature for 0.5 h, providing 5aa in 13% yield as the main product (entry 1). The structure was confirmed by single-crystal X-ray diffraction analysis and NMR spectra (Supporting inforamtion). Considering the influence of base on the reactivity, we firstly used KOtBu as the base. To our delight, the desired product was isolated in 41% yield of 5aa as well as 11% of 4aa (entry 2). This encouraging result prompted us to investigate other commonly used bases (entries 3–9). Na2CO3 was proved to be beneficial than various inorganic (Cs2CO3, K2CO3 and KHCO3) and organic bases (DMAP (N, N-dimethylpyridin-4-amine), DBU (2, 3, 4, 6, 7, 8, 9, 10-octahydropyr-imido[1, 2-a]azepine) and Et3N (triethylamine)), affording product 5aa in 85% yield (entry 3 vs. entries 2–9). Further screening solvent revealed that HFIP was proved to be optimal than others, such as toluene, MeCN, H2O, DCE, DCM and TFE (entries 10–15), vastly suppressed the reactivity. It might be the HFIP plays a positive effect on the azaoxyallyl cation intermediates [15]. Moreover, with reducing the loading of 1a or base, the yield of 5aa decreased obviously (entries 16 and 17). When the reaction temperature decreased to 0 ℃ from room temperature, the target product was obtained in 19% yield (entry 18). Finally, the optimized reaction conditions (entry 4) were determined as follows: N-(benzyloxy)-2-bromo-2-methylpropanamide (1a) (0.2 mmol), benzo[d]isoxazole 3a (0.1 mmol) and Na2CO3 (0.4 mmol) in HFIP (1 mL) at room temperature for 0.5 h.
With the optimized reaction conditions in hand, the applicability of this protocol was examined (Scheme 2). The effect of the substituents on the nitrogen atom of 1 was first examined. To our delight, electron-withdrawing or electron-donating groups (for example, -Me, -OMe, -Cl) on the phenyl ring at the aromatic alkoxy-substituents were well tolerated and gave the desired products in 53%–87% yields (5aa-5 da). Subsequently, we investigated aliphatic alkoxy-substituents to react with 3a, including -Me, -Et, -iPr, -tBu, -nBn, -allyl, giving the desired products (5ea-5 ja) in 57%–94% yield. Additionally, the reaction of N-phenyl-substituted 2-bromo-2-methylpropanamide 1k with 3a provided the corresponding product 5ka in 68% yield. However, the expected product (5la) was not detected for the substrate with benzyl on the nitrogen atom. Satisfyingly, cyclohexylsubstituted haloamide 1m reacted smoothly with 3a, giving the desired product 5ma in 75% yield. N-(Benzyloxy)-2, 2-dichloroacetamide 1n provided the unexpected product 5na in 14% yield. Other mono-substituted haloamide were also tried as reactants, e.g., monomethyl, monoethyl and monophenyl-substituted haloamide, no desired product was observed.
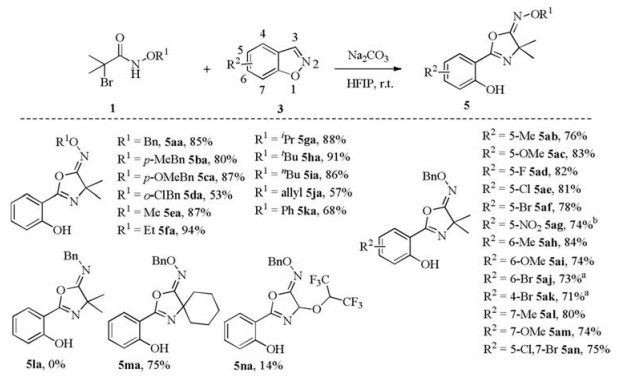
Furthermore, a range of benzo[d]isoxazoles were employed to probe the scope of the reaction. Satisfyingly, benzo[d]isoxazoles with electronic or steric effects groups proceeded smoothly with 2a to give the desired products (5ab-5an) in 71%–84% yields. Electron-donating (-Me, -OMe) and withdrawing (-F, -Cl, -Br) groups at 5-position on the benzo[d]isoxazoles were readily coupled with 2a to provide the corresponding products in 76%–83% yields (5ab-5af). We were delighted to find that the reaction of benzo[d]isoxazole with challenging groups (-NO2) also afforded the desired product (5ag) in 74% yield. These results indicated that the electron density of benzo[d]isoxazole did not significantly affect this transformation. The 6-substituted substrates also showed excellent compatibility with various functional groups, such as -CH3 (5ah), -OMe (5ai), -Br (5aj). In particular, substituents at both 4-and 7-position of 3 showed good reactivity with 71%–80% yields (5ak-5am), thus displaying tolerance of steric hindrance. Moreover, disubstituted benzo[d]isoxazole 3n worked well with 1a to provide the target product (5an) in 75% yield.
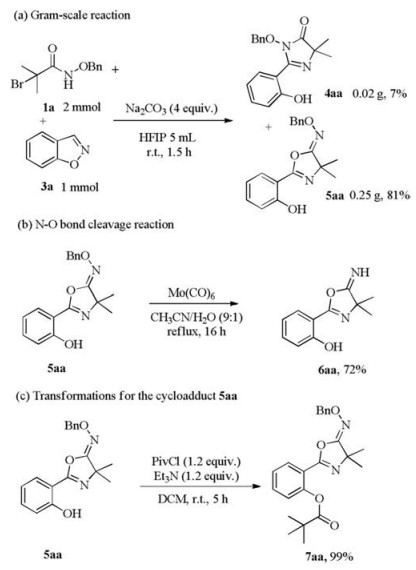
In order to showcase the robustness and practicality of this ring opening cycloaddition reaction, we carried out gram-scale reaction. As illustrated in Scheme 3a, 5aa and 4aa could be isolated in 0.25 g (81% yield) and 0.02 g (7% yield) under the optimized conditions, respectively. Cleavage of the N–O bond of 5aa could be readily achieved through refluxing with Mo(CO)6 in the mixture of MeCN/H2O (9:1), and 2-(5-imino-4, 4-dimethyl-4, 5-dihydrooxazol-2-yl)phenol 6aa was obtained in 72% yield (Scheme 3b). The hydroxyl group of 5aa could be converted to ester in the presence of PivCl (1.2 equiv) and Et3N (1.2 equiv.) at room temperature for 5 h. The tert-butoxycarbonyl (Boc) group protected product 7aa was obtained in 99% yield (Scheme 3c).
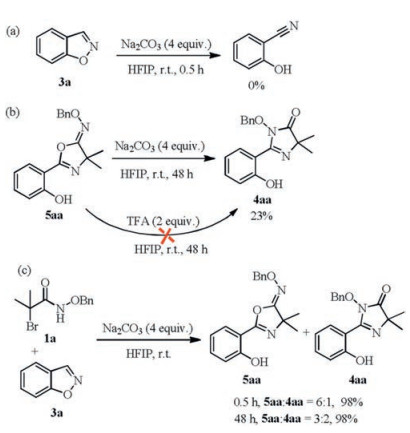
Some control experiments were investigated to clarify the reaction mechanism (Scheme 4). Initially, we wondered whether the cycloaddition reaction started from between salicylonitrile and azaoxyallylic cation. The model reaction was carried out under the optimal reaction conditions in the absence of 1a. The substrate 3a was not transformed to salicylonitrile (Scheme 4a). Then, when the product 5aa was treated with base or acid, the 2-hydroxyarylimidazolinone 4aa could be achieved in 23% yield with Na2CO3 and was not obtained with TFA, respectively (Scheme 4b). Additionally, the 2-hydroxyaryl-oxazolines 5aa was transformed to 2-hydroxyaryl-imidazolinone 4aa obviously by extending the reaction time from 0.5 h to 48 h under the optimal reaction conditions, indicating that the 2-hydroxyaryl-oxazolone 5aa may obtained as the stable kinetically preferred product, further could slowly rearrange to 4aa under certain conditions (Scheme 4c).
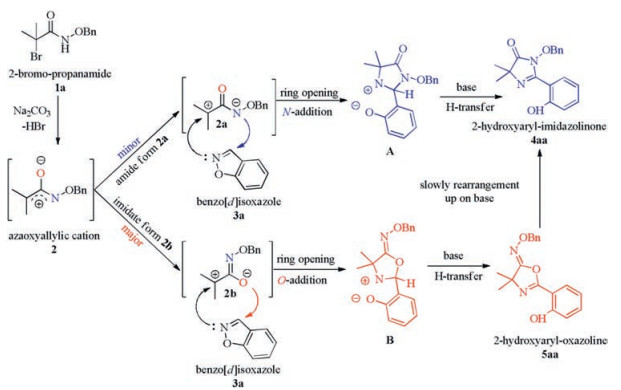
On the basis of the above results and previous reports, a plausible reaction mechanism is proposed in Scheme 5. Firstly, 2-bromo-propanamide 1a was in situ transferred to azaoxyallyl cation intermediate 2 under weakly basic conditions. The electrondonating group (-OBn) is indispensable to stabilize this intermediate. The ensuing cycloaddition of benzo[d]isoxazole 3a and azaoxyallyl cation 2 could occur through unevenly two routes. The minor tautomer amide form 2a of azaoxyallyl cation 2 exhibits the nitrogen atom as nucleophile delivers the N-cyclization intermediate A, following a H-transfer cascade reaction to afford the product 2-hydroxyaryl-imidazolinone 4aa. On the other hand, the major tautomer imidate form 2b of azaoxyallyl cation 2 through processes of O-addition and H-transfer produces the desired O-cyclization 2-hydroxyaryl-oxazoline 5aa. Additionally, due to their structure properties [9], the generated O-alkylated 5aa could slowly rearrange to 4aa under base condition.
In summary, we have reported a novel synthesis of 2-hydroxyaryl-oxazolines via regioselective ring-opening [3 + 2] cycloaddition reaction between azaoxyallyl cations and benzo[d]-isoxazoles, in which the imidate form intermediates (C~O 3-atom synthon) from azaoxyallyl cations were employed as the major cycloaddition intermediates. This procedure provided an efficient route to synthesize 2-hydroxyaryl-oxazolines in good yields under mild reaction conditions, exhibiting good functional group tolerance and gram-scale ability. Further applications of this methodology to the construction of natural products and pharmaceutical molecules are currently underway in our laboratory.
We greatly acknowledge partial financial support from the Ministry of Science and Technology of China (No. 2016YFE0132600), and Zhengzhou University.
Supplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.025.
(a) A. Padwa, W.H. Pearson, Synthetic Applications of 1, 3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, Hoboken, 2003;
(b) G. Pandey, P. Banerjee, S.R. Gadre, Chem. Rev. 106 (2006) 4484-4517;
(c) T. Hashimoto, Y. Takiguchi, K. Maruoka, J. Am. Chem. Soc.135 (2013) 11473-11476;
(d) H. Li, R.P. Hughes, J. Wu, J. Am. Chem. Soc. 136 (2014) 6288-6296;
(e) R. Narayan, M. Potowski, Z.J. Jia, et al., Acc. Chem. Res. 47 (2014) 1296-1310;
(f) M. Wang, Z. Huang, J. Xu, Y.R. Chi, J. Am. Chem. Soc. 136 (2014) 1214-1217;
(g) T. Hashimoto, K. Maruoka, Chem. Rev. 115 (2015) 5366-5412;
(h) H. Li, J. Wu, Synthesis 47 (2015) 22-33;
(i) H. Santos, A. Distiller, A.M. D'Souza, et al., Org. Chem. Front. 2 (2015) 705-712;
(j) S.I. Murahashi, Y. Imada, Chem. Rev. 119 (2019) 4684-4716.
(a) I. Lengyel, J.C. Sheehan, Angew. Chem. Int. Ed. 7 (1968) 25-36;
(b) Y. Kikugawa, M. Shimada, M. Kato, T. sakamoto, Chem. Pharm. Bull. 41 (1993) 2192-2194;
(c) K.L. Barnes, A.K. Koster, C.S. Jeffrey, Tetrahedron Lett. 55 (2014) 4690-4696.
J. Xuan, X. Cao, X. Cheng, Chem. Commun. 54(2018) 5154-5163. doi: 10.1039/C8CC00787J
(a) C.S. Jeffrey, K.L. Barnes, J.A. Eickhoff, et al., J. Am. Chem. Soc. 133 (2011) 7688-7691;
(b) C.S. Jeffrey, A. Acharya, J. Eickhoff, Synthesis 45 (2013) 1825-1836.
(a) Y. An, H. Xia, J. Wu, Chem. Commun. 52 (2016) 10415-10418;
(b)Q.Jia, D.Li, M.Lang, K.Zhang, J.Wang, Adv.Synth.Catal.359 (2017)3837-3842;
(c) G. Wang, R. Chen, M. Wu, et al., Tetrahedron Lett. 58 (2017) 847-850;
(d) G. Wang, S. Zhao, R. Chen, et al., Tetrahedron Lett. 58 (2017) 4308-4311;
(e) J. Xuan, X. Cheng, X. Cao, ChemistrySelect 2 (2017) 4364-4367;
(f) H.W. Zhao, Y.D. Zhao, Y.Y. Liu, et al., Eur. J. Org. Chem. 24 (2017) 3466-3472;
(g) H.W. Zhao, Y.D. Zhao, Y.Y. Liu, et al., RSC Adv. 7 (2017) 12916-12922;
(h) B. Balde, G. Force, L. Marin, et al., Org. Lett. 20 (2018) 7405-7409;
(i) X. Cheng, X. Cao, J. Xuan, W.J. Xiao, Org. Lett. 20 (2017) 52-55;
(j) M.C. DiPoto, J. Wu, Org. Lett. 20 (2018) 499-501;
(k) D. Ji, J. Sun, Org. Lett. 20 (2018) 2745-2748;
(l) R. Singh, K. Nagesh, D. Yugandhar, et al., Org. Lett. 20 (2018) 4848-4853;
(m) G. Wang, R. Chen, S. Zhao, et al., Tetrahedron Lett. 59 (2018) 2018-2020;
(n) X. Xu, K. Zhang, P. Li, H. Yao, A. Lin, Org. Lett. 20 (2018) 1781-1784;
(o) Y. Zhang, H. Ma, X. Liu, et al., Org. Biomol. Chem. 16 (2018) 4439-4442;
(p) X. Cheng, X. Cao, S.J. Zhou, et al., Adv. Synth. Catal. 361 (2019) 1230-1235;
(q) Z.L. He, Y. Chen, X. Wang, M. Ni, G. Wang, Tetraherdron 75 (2019) 130461.
(a) A. Acharya, D. Anumandla, C.S. Jeffrey, J. Am. Chem. Soc.137 (2015) 14858-14860;
(b) M.C. DiPoto, R.P. Hughes, J. Wu, J. Am. Chem. Soc.137 (2015) 14861-14864;
(c) W. Ji, L. Yao, X. Liao, Org. Lett. 18 (2016) 628-630;
(d) K. Zhang, X. Xu, J. Zheng, et al., Org. Lett. 19 (2017) 2596-2599.
(a) A. Acharya, K. Montes, C.S. Jeffrey, Org. Lett. 18 (2016) 6082-6085;
(b) K. Zhang, C. Yang, H. Yao, A. Lin, Org. Lett. 18 (2016) 4618-4621;
(c) Q. Jia, Z. Du, K. Zhang, J. Wang, Org. Chem. Front. 4 (2017) 91-94;
(d) S. Jiang, K. Li, J. Yan, et al., J. Org. Chem. 82 (2017) 9779-9785;
(e) P.L. Shao, Z.R. Li, Z.P. Wang, et al., J. Org. Chem. 82 (2017) 10680-10686;
(f) S.J. Zhou, X. Cheng, J. Xuan, Asian J. Org. Chem. 8 (2019) 1-5;
(g) V. Jaiswal, B. Mondal, K. Singh, et al., Org. Lett. 21 (2019) 5848-5852.
C. Li, K. Jiang, Q. Ouyang, T.Y. Liu, Y.C. Chen, Org. Lett. 18(2016) 2738-2741. doi: 10.1021/acs.orglett.6b01194
(a) T. Keumi, T. Morita, K. Teramoto, et al., J. Org. Chem. 51 (1986) 3439-3446;
(b) H. Maruoka, F. Okabe, K.J. Yamagata, Heterocycles 74 (2007) 383-396;
(c) J. Yuan, C.B. Rao, Y. Liang, et al., Adv. Synth. Catal. 361 (2019) 160-169.
(a) X. Lei, M. Gao, Y. Tang, Org. Lett. 18 (2016) 4990-4993;
(b) Y.P. Han, X.S. Li, Z. Sun, et al., Adv. Synth. Catal. 359 (2017) 2735-2740;
(c) Z. Chen, C. Han, C. Fan, G. Liu, S. Pu, ACS Omega 3 (2018) 8160-8168;
(d) Y.B. Pandit, R.L. Sahani, R.S. Liu, Org. Lett. 20 (2018) 6655-6658.
(a) C.G. Marshall, M.D. Burkart, T.A. Keating, et al., Biochemistry 40 (2001) 10655-10663;
(b) R. Bergeron, N. Bharti, S. Singh, Synthesis 7 (2007) 1033-1037;
(c)A.Sakakura, S.Umemura, R.Kondo, etal., Adv.Synth.Catal.349 (2007)551-555;
(d) P. Fu, P. Liu, H. Qu, et al., J. Nat. Prod. 74 (2011) 2219-2223;
(e) N. Liu, F. Shang, L. Xi, Y. Huang, Mar. Drugs 11 (2013) 1524-1533;
(f) W.J. Wu, J.W. Zhang, W.J. Zhang, S.P. Wei, Heterocycles 89 (2014) 1656-1661;
(g) C. Ghosh, S. Pal, A. Patel, et al., Org. Lett. 20 (2018) 6511-6515.
(a) D.S.C. Black, M. Wade, Aust. J. Chem. 25 (1972) 1797-1810;
(b) H. Yang, M.A. Khan, K.M. Nicholas, Organometallics 12 (1993) 3485-3494;
(c) S. Rajaram, M.S. Sigman, Org. Lett. 4 (2002) 3399-3401;
(d) D. Franco, M. Gómez, F. Jiménez, et al., Organometallics 23 (2004) 3197-3209;
(e) J.X. Qiao, T.C. Wang, H. Carol, et al., Org. Lett.13 (2011) 1804-1807;
(f) W. Dan, H. Geng, J. Qiao, et al., Molecules 21 (2016) 96.
(a) T. Yuan, C. Pi, C. You, et al., Chem. Commun. 55 (2018) 163-166;
(b) S. Du, C. Pi, T. Wan, Y. Wu, X. Cui, Adv. Synth. Catal. 361 (2019) 1766-1770;
(c) J. Ren, C. Pi, Y. Wu, X. Cui, Org. Lett. 21 (2019) 4067-4071;
(d) Z. Shen, C. Pi, X. Cui, Y. Wu, Chin. Chem. Lett. 30 (2019) 1374-1378;
(e) Z. Yang, C. Pi, X. Cui, Y. Wu, Org. Chem. Front. 6 (2019) 2897-2901;
(f) L. Xu, L. Wang, Y. Feng, et al., Org. Lett. 19 (2017) 4343-4346;
(g) Y. Li, C. Jia, H. Li, et al., Org. Lett. 20 (2018) 4930-4933;
(h) Z. Yang, L. Jie, Z. Yao, X. Yang, X. Cui, Adv. Synth. Catal. 361 (2019) 214-218;
(i) L. Wang, D. Xiong, L. Jie, C. Yu, X. Cui, Chin. Chem. Lett. 29 (2018) 907-910.
J. Feng, M. Zhao, X. Lin, J. Org. Chem. 84 (2019) 9548-9560. doi: 10.1021/acs.joc.9b01166
(a) M. Harmata, C. Huang, P. Rooshenas, P.R. Schreiner, Angew. Chem. Int. Ed. 47 (2008) 8696-8699;
(b) A.G. Myers, J.K. Barbay, Org. Lett. 3 (2001) 425-428.

Scheme 2 Extension of reaction scope. Reactions were carried out with 0.2 mmol of 1 and 0.1 mmol of 3 in the presence of 0.4 mmol of Na2CO3 in 1.0 mL of HFIP at room temperature for 0.5 h. Isolated yield. a 1 h. b 2 h.

Scheme 3 Further study on [3 + 2] cycloaddition reaction between azaoxyallyl cations and benzo[d]isoxazoles.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

