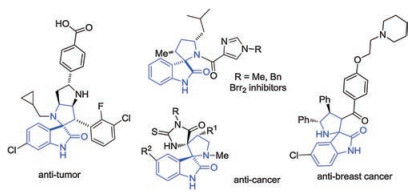
Figure 1.
Examples of biological active spiro[pyrrolidin-3, 2'-oxindoles].
Remote regioselective organocatalytic asymmetric [3+2] cycloaddition of N-2, 2, 2-trifluoroethyl isatin ketimines with cyclic 2, 4-dienones
Chuncheng Zou , Yuanyuan Han , Chuikun Zeng , Tony Y. Zhang , Jinxing Ye , Gonghua Song

As commonly known, spirooxindoles, especially spiro[pyrrolidin-3, 2'-oxindoles] are one of the most important synthetic targets due to their remarkable biological activities and stable structures compared with spiro[pyrrolidin-3, 3'-oxindoles] (Fig. 1) [1]. The challenge of simultaneous construction of spiro quaternary carbon centers and multiple chiral centers has attracted the attention of organic chemists in the past few years. A plethora of enantioselective methodologies for synthesis of these complex structures have been developed including ringclosing metathesis reaction [2], rearrangement [3] and cycloaddition strategy [4]. Among these different well-developed methods, the catalytic asymmetric 1, 3-dipolar cycloaddition between azomethine ylide and activated alkene has been the most prevalent and powerful tool [5]. On the other hand, the corporation of trifluoromethyl into bioactive molecules can bring positive effects, such as enhancing binding affinity to molecular receptors in the development of novel pharmaceuticals and agrochemicals [6]. Recently, N-2, 2, 2-trifluoroethyl isatin ketimines [7] have served as efficient azomethine ylide precursors to generate CF3-containing 3, 2'-spiro[pyrrolidin-3, 2'-oxindoles] with active alkenes via [3 + 2] cycloaddition. In 2015, Wang group reported the asymmetric synthesis of the spiro[pyrrolidin-3, 2'-oxindoles] via an organocatalytic 1, 3-dipolar cycloaddition with nitrostyrenes [7a] or enals [7b] (Scheme 1, a and b). Later, Yuan and co-workers developed a thiourea-tertiary amine catalyzed asymmetric [3 + 2] cycloaddition reaction of N-2, 2, 2-trifluoroethyl isatin ketimines and 3-alkenyl-5-arylfuran-2(3H)-ones for construction of chiral spiro[pyrrolidin-3, 2'-oxindoles] (Scheme 1, c) [7d]. Followed that, Enders and Lu groups disclosed separately Michael–Mannich [3 + 2] cycloaddition between isatin ketimines and isatin-derived enolates employing bifunctional catalysts [7f, 7g] (Scheme 1, d). However, those achievements are limited to α, β-activated alkenes. Those elegant achievements provided meaningful insights for the remote regioselective catalytic functionalization of activated alkenes.
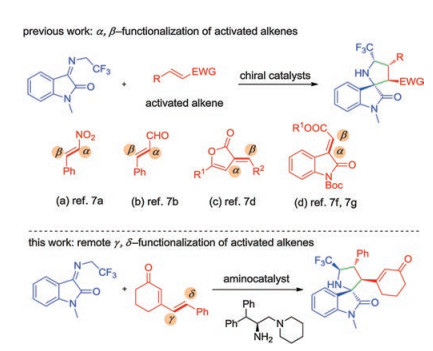
For cyclic 2, 4-dienone that have multiple reaction sites [8] under the catalysis of organocatalyst, selective activation of the γ, δ-alkene was a challenge. In 2013, Melchiorre and co-workers used cascade strategy to realize γ, δ-functionalization of cyclic 2, 4-dienones that acted as an electron donor-acceptor [9]. After that, they reported a chiral phosphoric acid catalytic vinylogous [4 + 2] cycloaddition of cyclic 2, 4-dienones with 2-vinylindoles [10].
To the best of our knowledge, remote regioselective γ, δfunctionalization of cyclic 2, 4-dienones with N-2, 2, 2-trifluoroethyl isatin ketimines as dipolarophile via [3 + 2] cycloaddition still remains a challenging task. Herein, encouraged by these achievements and our research in this field, we presented an organocatalytic asymmetric [3 + 2] cycloaddition reaction of trifluoromethyl-containing ylides with cyclic 2, 4-enones (Scheme 1, bottom).
Key to achieve the designed asymmetric [3 + 2] cycloaddition between trifluoromethyl-containing azomethine ylides and cyclic 2, 4-dienones was the ability of chiral organocatalysts to activate the remote γ, δ-alkene. Then, a series of organic primary amine catalysts were tested, using 1a as 1, 3-dipole and 2a as the model reactants, 40 mol% of benzoic acid as co-catalyst at 0 ℃ for 24 h. Fortunately, catalyzed by the cinchona-based primary amine 3a, the desired γ, δ-regioselective [3 + 2] product 4a was provided with excellent diastereoselectivity and poor enantioselectivity. The utilization of quinine derived thiourea bifunctional organocatalyst 3b also showed the similar result. Based on our previous experience on the flexible diamine catalysts, we then tested the catalytic performance of primary-secondary or primary-tertiary diamine catalysts 3c-3f (Table 1, entries 3–6). We were delighted to find that the yield and enantioselectivity were greatly improved under the catalysis of 3f even with decreasing diastereoselectivity. Then, 3f was employed as the catalyst to screen other factors, such as solvents, temperature and additive acids. The evaluation of solvents revealed that chloroform induced an appreciable level of enantioselectivity but lower diastereoselectivity and yield. Other solvents, such as toluene, THF and DCE also decreased the stereoselectivites (Supporting information). We found that the temperature greatly affected the reactivity and stereoselectivites. Running the reaction at -20 ℃ further increased the diastereoselectivity and enantioselectivity with the slightly decreasing yield. While only 11% yield was obtained at -40 ℃ even with better stereoselectivity. After screening a variety of acids, 4-methylbenzoic acid provided outstanding diastereoselectivity while maintaining higher yield and enantioselectivity. Finally, the excellent yield and stereoselectivity were achieved with the use of 40 mol% 4-methylbenzoic acid, 20 mol% 3f and DCM as solvent at -20 ℃ for 48 h (83% yield, 90% ee and 15:1 dr). It was available to scale up the reaction, and 1.69 g of 4a (77% yield, 87% ee and 14:1 dr) were obtained.
With the optimal reaction conditions in hand (Table 1, entry 11), the scope of N-2, 2, 2-trifluoroethyl isatin ketimines for the catalytic asymmetric [3 + 2] cycloaddition reaction was then evaluated (Scheme 2). The ketimines bearing with different N-substituents were investigated, such as ethyl and benzyl groups, and reveals excellent yields and good diastereoselectivity and enantioselectivity (Scheme 2, 4c, 4d). But, due to poor solubility in DCM, N-H substituted ketimine transformed into the desired product in poor yield and decreasing stereocontrol (Scheme 2, 4b).
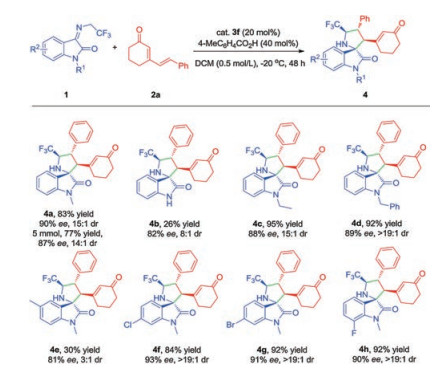
Next, we investigated the effect of substituents on the indolinone ring. 5-Methyl substituted ketimines revealed poor reactivity and moderate enantio-and diastereoselectivity (Scheme 2, 4e). While, in the presence of halogen atoms in the 6 or 7-position, the process performed with excellent yields and diastereoselectivity as well as satisfying enantioselectivity (Scheme 2, 4f-4g).
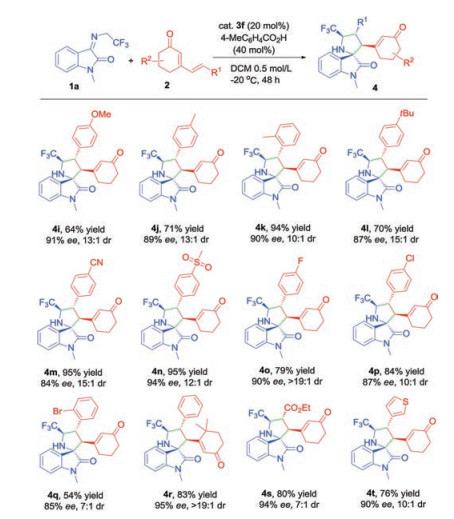
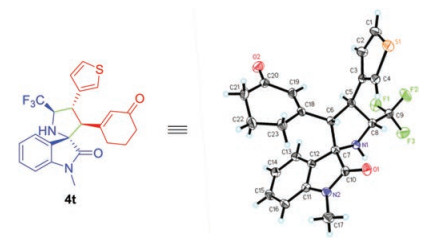
We further examined the generality of the asymmetric [3 + 2] cycloaddition to various cyclic 2, 4-dienones. As shown in Scheme 3, a wide range of cyclic 2, 4-dienones with different electronic and structural substituents were well tolerated in this transformation. For electron-donating groups (Me, MeO and tBu) substituted dienones at δ-position of aromatic ring, the cycloaddition proceeded smoothly and provided corresponding products in good to excellent yields and high stereoselectivities (Scheme 3, 4i, 4j, 4l). With respect to the electron-withdrawing groups, such as cyano group, sulfonyl group and halogens, similar results were obtained (4m-4q). Bulky reactant with gem-dimethyl group on the cyclohexanone underwent the conversion efficiently, delivering 4r in 83% yield and excellent stereocontrol (95% ee, >19:1 dr). Remarkably, when the aromatic moiety was replaced by ester group, the cycloaddition also revealed good yield and excellent enantioselectivity, albeit with moderate diastereoselectivity (4s). The δ-thiofuran substituted cyclic 2, 4-dienone was also the suitable dipolarophile and constructed enantioenriched heteroaryl derivative 4t. The absolute configuration of 4t was determined by X-ray crystallographic analysis (Fig. 2).
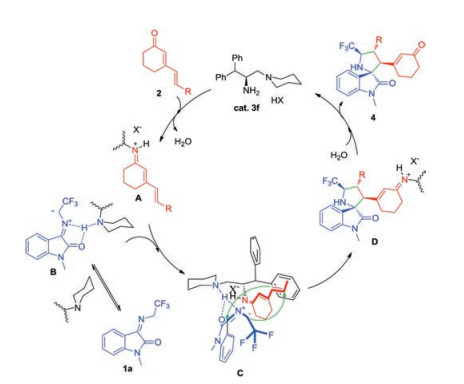
Combining previous studies on dipolar cycloaddition reactions [7a, 7b, 7g], we proposed a possible mechanism as shown in Scheme 4. Firstly, the reaction of primary amine with cyclic 1, 4-dienone 2 gave the intermediate iminium ion A. Concurrently, the N-2, 2, 2-trifluoroethylisatin ketimine was activated via the Hbonding interactions with the protonated tertiary amine of cat. 3f and afford the intermediate C with enhanced nucleophilicity of the reacting carbon anion. Subsequently, [3 + 2] cycloaddition reaction gave iminium ion D. Finally, D was hydrolyzed to give the final product 4, and the primary-tertiary amine catalyst was regenerated.
In conclusion, we have presented an asymmetric [3 + 2] cycloaddition reaction of trifluoromethyl-containing ylides and cyclic 2, 4-dienones by using the simple organic primary-tertiaryamine catalyst. With this newly developed method, a series of functionalized spiro [pyrrolidin-3, 2'-oxindoles], bearing a trifluoromethyl group with remarkable biological activities, could be directly accessed in a scalable fashion with good to excellent yields and high stereoselectivities.
This work was supported by the National Natural Science Foundation of China (Nos. 21272068, 21572056, 21801077), China Postdoctoral Science Foundation (Nos. 2017M621382, 2018T110355), Shanghai Sailing Program (No. 18YF1406200), and the Fundamental Research Funds for the Central Universities.
Supplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.052.
(a) A. Gollner, D. Rudolph, H. Arnhof, et al., J. Med. Chem 59 (2016) 10147-10162;
(b) M. Ito, M. Iwatani, T. Yamamoto, et al., Bioorg. Med. Chem 25 (2017) 4753-4767;
(c) Y.A. Ivanenkov, S.V. Vasilevski, E.K. Beloglazkina, et al., Bioorg. Med. Chem. Lett. 25 (2015) 404-409;
(d) A. Kumar, G. Gupta, A.K. Bishnoi, et al., Bioorg. Med. Chem. 23 (2015) 839-848.
(a) B. Alcaide, P. Almendros, R. Rodriguez-Acebes, J. Org. Chem. 71 (2006) 2346-2351;
(b) G. Lesma, N. Landoni, A. Sacchetti, A. Silvani, Tetrahedron 66 (2010) 4474-4478;
(c) K. Jiang, Z.J. Jia, S. Chen, L. Wu, Y.C. Chen, Chem. -Eur. J. 16 (2010) 2852-2856;
(d) M.P. Castaldi, D.M. Troast, J.A. Porco Jr, Org. Lett. 11 (2009) 3362-3365.
(a) U.K.S. Kumar, H. Ila, H. Junjappa, Org. Lett. 3 (2001) 4193-4196;
(b) Z. Szakonyi, F. Fülöp, D. Tourwé, N.D. Kimpe, J. Org. Chem. 67 (2002) 2192-2196;
(c) E. Zhang, C.A. Fan, Y.Q. Tu, F.M. Zhang, Y.L. Song, J. Am. Chem. Soc.131 (2009) 14626-14627.
(a) R. Rios, Chem. Soc. Rev. 41 (2012) 1060-1074;
(b) G.S. Singh, Z.Y. Desta, Chem. Rev. 112 (2012) 6104-6155;
(c) D. Cheng, Y. Ishihara, B. Tan, C.F. Barbas III, ACS Catal. 4 (2014) 743-762.
(a) M. Bonin, A. Chauveau, L. Micouin, Synlett (2006) 2349-2363;
(b) T.M.V.D. Pinho e Melo, Eur. J. Org. Chem (2006) (2006) 2873-2888;
(c) G. Pandey, P. Banerjee, S.R. Gadre, Chem. Rev 106 (2006) 4484-4517;
(d) H. Pellissier, Tetrahedron 63 (2007) 3235-3285;
(e) L.M. Stanley, M.P. Sibi, Chem. Rev 108 (2008) 2887-2902;
(f) C. Nájera, J.M. Sansano, Top. Heterocycl. Chem. 12 (2008) 117-145;
(g) J. Adrio, J.C. Carretero, Chem. Commun 47 (2011) 6784-6794;
(h) J. Adrio, J.C. Carretero, Chem. Commun 50 (2014) 12434-12446;
(i) T. Hashimoto, K. Maruoka, Chem. Rev 115 (2015) 5366-5412.
(a) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, WileyBlackwell, Chichester, 2009;
(b) V.A. Petrov, Fluorinated Heterocyclic Compounds: Synthesis Chemistry and Application, Wiley, Hoboken, 2009;
(c) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd, Wiley-VCH, Weinheim, 2013.
(a) Q. Sun, X. Li, J. Su, et al., Adv. Synth. Catal. 357 (2015) 3187-3196;
(b) M. Ma, Y. Zhu, Q. Sun, et al., Chem. Commn. 51 (2015) 8789-8792;
(c) X. Li, J. Sun, Z. Liu, et al., Org. Lett. 18 (2016) 956-959;
(d) Z.H. Wang, Z.J. Wu, D.F. Yue, et al., Chem. Commun. 52 (2016) 11708-11711;
(e) M. Shi, Y.N. Gao, Eur. J. Org. Chem. (2017) (2017) 1552-1560;
(f) Y. Zhi, K. Zhao, C. Essen, K. Rissanen, D. Enders, Synlett 28 (2017) 2876-2880;
(g) W.J. Huang, Q. Chen, N. Lin, et al., Org. Chem. Front. 4 (2017) 472-482;
(h) A. Ponce, I. Alonso, J. Adrio, J.C. Carretero, Chem. -Eur. J. 22 (2016) 4952-4959;
(i) W.J. Huang, Q. Chen, W.R. Zhu, et al., Heterocycles 94 (2017) 879-893.
(a) X. Tan, Y. Liu, P. Melchiorre, Angew. Chem. Int. Ed 51 (2012) 6439-6442;
(b) T. Kitanosono, P. Xu, S. Kobayashi, Chem. Commun. 49 (2013) 8184-8186;
(c) T. Kitanosono, P. Xu, S. Kobayashi, Chem. -Asian J. 9 (2014) 179-188;
(d) X. Gu, T. Guo, Y. Dai, et al., Angew. Chem. Int. Ed 54 (2015) 10249-10253;
(e) Y. Wei, Z. Liu, X. Wu, et al., Chem. -Eur. J. 21 (2015) 18921-18924;
(f) C. Zou, C. Zeng, Z. Liu, et al., Angew. Chem. Int. Ed. 55 (2016) 14257-14261;
(g) X. Sun, J. Fei, C. Zou, M. Lu, J. Ye, RSC Adv. 6 (2016) 106676-106679;
(h) W. Xiao, Q.Q. Yang, Z. Chen, et al., Org. Lett. 20 (2018) 236-239.
X. Tan, P. Melchiorre, Angew. Chem. Int. Ed. 52 (2013) 5360-5363. doi: 10.1002/anie.201301017
X. Tan, N. Hofmann, P. Melchiorre, Angew. Chem. Int. Ed 53 (2014) 2997-3000. doi: 10.1002/anie.201310487

Scheme 2 Substrate scope of substituted N-2, 2, 2-trifluoroethyl isatin ketimines. All reactions were performed under the optimized reaction conditions. Isolated yields of products were given. The ee values were determined by chiral HPLC analysis. The dr values were determined by 1H NMR analysis of the crude reaction mixtures.

Scheme 3 Substrate scope of substituted cyclic dienones. All reactions were performed over 48 h with 1a (0.1 mmol, 0.5 mol/L), 2 (0.15 mmol), and cat. 3f (20 mol%), 4-MeC6H4CO2H (40 mol%) in DCM at -20 ℃. Isolated yields of products were given. The ee values were determined by chiral HPLC analysis. The dr values were determined by 1H NMR analysis of the crude reaction mixtures.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:


