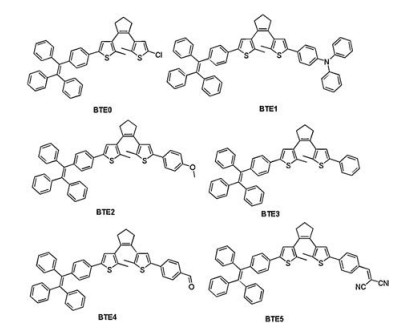
Figure 1.
Chemical structures of the BTE compounds.
Unsymmetrical photochromic bithienylethene-bridge tetraphenylethene molecular switches: Synthesis, aggregation-induced emission and information storage
Liangwei Ma , Chengpeng Li , Qing Yan , Sheng Wang , Wangen Miao , Derong Cao

Diarylethene (DAE), a family of classic P-type photochromic molecules, can be reversibly transformed between closed-ring and open-ring forms by photo irradiation, accompanying with significantly changes of polarizability, color, conformation, and spectroscopic property [1, 2]. These compounds have been extensively applied in functional materials for their excellent stability and fatigue resistance. Particularly, the elaboration of molecular systems combining DAEs and fluorescence properties constituted a promising research field for their potential in sensor [3], information storage [4], anti-counterfeiting [5], cell image [6] and super-resolution imaging [7-11] for their high sensitivity, resolution, contrast, and fast response times of the fluorescence especially the remarkable fatigue resistance.
However, normal fluorophores suffer from aggregation-caused quenching (ACQ) phenomenon which is caused by the strong intermolecular π-π interaction or hydrogen bonding between neighboring fluorophores in aggregation state generally [12-14]. Their fluorescences are debased or quenched in the aggregation state, limiting their applications greatly. Fortunately, a novel class of fluorophores with aggregation-induced emission (AIE) characteristic, opposite to ACQ phenomenon, has been discovered, providing a new direction to design organic fluorescent materials with more practical promising applications [15]. Attracted by the fascinating phenomenon and promising applications, many research groups have enthusiastically engaged in AIE studies [6, 16-24].
Despite several DAE derivatives with AIE property have been reported, there is still room for improvement and there is a need for research that will enable understanding for the rational design of DAE with promising photochromic behavior and AIE property, particularly those derivatives could be easily synthesized and derived [13, 25-28].
Recently, tetraphenylethene (TPE) has attracted increasing attention for its excellent thermal property, high fluorescence quantum yield and excellent AIE property [29, 30]. In our previous work, we designed unilateral and bilateral TPE substituted BTE derivatives [31]. Those compounds exhibited both AIE and photochromic properties on the same time. With our continuous interest in designing various AIE-active photochromic materials, we proposed that further grafted other building block on the other side of unilateral TPE substituted bithienylethene (BTE0) would offer a strategy to design new AIE-active photochromic system. Herein, we report the synthesis and characterization, as well as the photophysical and photochromic properties of unsymmetrical photochromic BTE-bridge derivatives (Fig. 1). The present work provides important insights and guiding principles into the molecular design of unsymmetrical photochromic bithienylethene systems with excellent AIE properties.
Photochromic properties of those compounds were investigated by UV–vis spectroscopy. All the compounds demonstrated good solubility in typical organic solvents such as THF, acetonitrile, CH2Cl2, and ethyl acetate, but they were hardly soluble in water. BTE1–5 all exhibited quite similar absorption spectra in THF solutions as shown in Fig. 2a and Fig. S16 (Supporting information). They all aroused a sharp absorption peak at 330–350 nm which may attribute to the absorption of TPE segments. Another peak at 280–300 nm of those dyads except BTE5 should belong to the absorption of bithienylethene section [30]. Indeed, BTE5 exhibited a small absorbance peak around 290 nm. However the peak around 290 nm was merged with the peak at 338 nm due to the weak absorbance of the prior peak. Upon irradiation of 365 nm light, the THF solutions of the open forms of those photochromic bithienylethene derivatives (BTE1–5) were found to exhibit color change from colorless or pale yellow solutions to purple, amaranthine, bluish or mint green. And all the peaks of those dyads at 330– 350 nm and 280–300 nm regions were gradually decreased in absorbance, except the peak at 300 nm of BTE4 was increased and a newabsorptionpeak at 554, 540, 542, 572 and 686 nm, respectively, were aroused upon irradiation of 365 nm light (Fig. 2a and Fig. S16). In addition, a clear isosbestic point were observed at 374, 368, 369, 379, 396 nm, respectively. These phenomena jointly indicated the occurrence of photocyclization reactionand formation of the closed forms [2]. Then irradiation those solutions with visible light, the color gradually faded and the absorption spectra recovered to original state gradually.
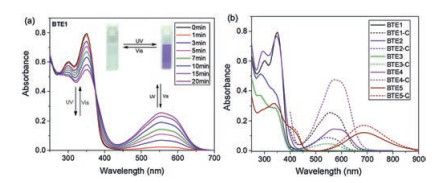
As shown in Fig. 2b, BTE with different substituent shown different photochromic absorbance peaks in THF solution. The open form of those dyads exhibited similar absorption spectra, except BTE5 extended to visible region. However, the closed form dyads shown significant different spectra. Except BTE1, the maximum absorption peaks were red-shifted with the decreased of electron-donating ability or the increased of electron-withdrawing ability of the substituent. For those dyads in open ring form, π-conjugation was localized in each thiophene ring and correspondingly substituents, and the spectra were similar to a substituted thiophene. But when converted to closed ring form by UV light irradiation, the π-conjugation delocalizes throughout the whole molecule. The modification of the substituent on the side of BTE bridges could impact the conjugation system significantly. Thereby, the maximum absorption wavelength of the closed ring form of BTE2-5 were shifted follow the electron-donating ability or the increased of electron-withdrawing property. However, BTE1, with triphenylamine as substitute, was red-shifted compared to BTE2 and BTE3, which was substituted by metoxybenzene and phenyl, respectively. Although, the electron-donating ability of triphenylamine substitute is larger than metoxybenzene and phenyl, the triphenylamine building block extend the conjugation configuration, and this may induce the red-shift of the absorption peak compared to BTE2 and BTE3. Besides, the cyclization quantum yields of BTE1-5 were determined to be 0.21, 0.02, 0.04, 0.44, 0.31, respectively [32, 33]. The quantum yields could be affected by the electron-withdrawing ability of side substituent greatly. A appropriate electron-withdrawing substituent is the key to obtain high cyclization quantum yields materials.
Fluorescent properties of those compounds were investigated by fluorescent spectroscopy. BTE1–5 shown strong emission when excited by 300 nm light. And the emission peak red-shifted along with the increase of electron-withdrawing ability of substituent group. Typically, emission peaks were red-shifted from 449 nm to 562 nm when triphenylamine substituent was replaced by 2-benzylidenemalononitrile. Emission spectra of all those compounds in THF and THF/water solutions were recorded to investigate their AIE or AIEE properties. The luminogens were able to aggregate in the aqueous mixtures with high water fractions (fw) due to their water insoluble character. Their fluorescence (FL) spectra and the changes of FL intensity with different fw were shown in Fig. 3 and Fig. S17 (Supporting information).
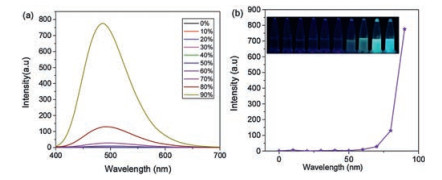
It is worth noting that BTE1 was obtained by decorating BTE core with a typical AIE element (TPE) and a traditional ACQ luminophore (triphenylamine). As shown in Fig. 3, the THF solution of BTE1 was almost none emission, which means the multiple phenyl rings on TPE moieties still could effectively dissipating the excited-state energy via intramolecular rotations even the two luminophores were connected through a BTE bridge when BTE1 was molecularly dissolved. With the water fraction increased, the molecule aggregated gradually accompanied with the fluorescence enhancement. And the fluorescence intensity increased to maximum value with the water fraction increased it to 90%. We believed this phenomenon was result from the propeller shaped TPE moieties increased the structural bulk and nonplanarity of the whole molecule and hence prevented π-π stacking interactions from forming in the aggregate state, and as a result, the intermolecular steric effects block the non-radiative decay channels resulting in the drastically fluorescence enhancement. These results jointly indicated ACQ phores could be converted to AIE luminophores by grafting AIE elements the ACQ core via BTE-bridge, providing a new strategy to the molecular design of photochromic bithienylethene systems with excellent AIE property.
BTE2–4 also exhibited typically AIE properties like BTE1. As shown in Fig. S17, only extremely weak signals were recorded in fluorescence spectra for those three dyads in THF solutions. Obvious fluorescence intensity change could not be observed until the fw up to 60% or 70%. The intensity gradually increased with the fw enhanced to 80%. Further increased to 90%, the intensity sharply boosted to maximum value. In addition, clearly fluorescence change could be identified by naked eyes under 365 nm UV light. As shown in the inset picture of Fig. S17, clear fluorescence emission could be observed only when the fw over 50% or 60%. And with the further increase of fw, the emission intensity was enhanced rapidly. These phenomena were corresponding with the fluorescence spectra. The phenyl rings of TPE segment in those dyads in dilute THF or low water contents (fw ≤ 50%) THF solutions could undergo active intramolecular rotations and vibrations, which serves as a relaxation channel for the excited states to nonradiative decay to the ground state. And this induced the extremely weak emission of those solutions. With the increased of fw, the molecule aggregated gradually for their water insoluble characters. And the intramolecular rotations were restricted due to the involved physical constraint, which blocks the non-radiative relaxation channel and opens the radiative decay pathway, which may explain the fluorescence enhancement behavior [29].
Different to the other four dyads, BTE5 exhibited AIEE characteristic. As described in Fig. S17, BTE5 emits an orange light at 577 nm in THF solution. With a small amount water was added, the emission was red-shift and catastrophically weakened when a small amount of water (10%) was added into THF, owing to the increase in solvent polarity and the transformation to twisted intramolecular charge transfer (TICT) state which was often observed in D-A system molecular [16]. The fluorescence keep decreased and remain invisible with the fw increased. When the fw increased to 60%, the fluorescence was revitalized, further increased and blue-shifted with the increased of fw. In the aqueous mixtures with high water contents (fw ≤ 60%), the fluorophore clustered together to form microaggregates. The hydrophobic environment and the restriction to intramolecular motions in the aggregates lead to the observed blue shift in the emission color and enhancement in the emission intensity.
In addition, the maximum emission wavelength of BTE1–4 were at 485, 489, 488 and 487 nm, respectively. The electrondonating or normal electron-withdrawing substituent seems impact slightly on the emission of TPE segment, which may result from the weak conjugation of two thiophene segment. However, when the substituent was replaced by a strong electronwithdrawing building block, the dyad was converted to an AIEE molecule which may undergo TICT mechanism. And the emission was red-shifted to 557 nm.
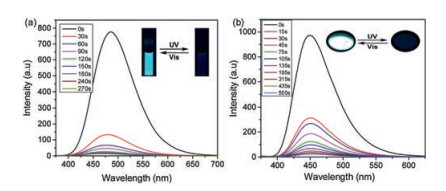
As shown in Fig. 2b, the closed form of BTE1–5 aroused a newly broad absorption peak on the regions of 450–650, 425–650, 425– 650, 450–700, 500–900 nm, respectively. Meanwhile, the maximum emission peaks of those dyads were at 485, 489, 488, 487 and 557 nm, respectively (Fig. 3 and Fig. S17), which were overlapped with the absorption peaks pretty well. Therefore, the emission of closed form dyads might be quenched for the energy transfer channel from TPE segment to BTE section could be opened. As we speculated, the emission of those dyads in 90% H2O/THF mixed solutions were faded gradually when exposed to 365 nm UV light (Fig. 4 and Fig. S18 in Supporting information). And the emission will revitalize and recover to the initial state up visible light irradiation for the photo-induced cycloreversion reaction blocked the energy transfer process by convert the accepter. Similar to those solutions, BTE1–5 exhibited the similar fluorescence "ON" and "OFF" process on solid state for the photo-induced cyclization and cycloreversion reaction could also occur and block or unblock the energy transfer process in solid state [27, 34].
In order to further understand the AIE properties of BTE1, FESEM images of their aggregates were studied. The optimized fw for BTE1 in mixed THF/H2O solution are 90%. Therefore, during the aggregation process, we investigated the influence of water contents (fw = 30%, 60% and 90%) on aggregate formation with BTE1. As shown in Fig. S19 (Supporting information), by increasing the water content from 30% to 60% and 90%, the morphology of BTE1 aggregates changed in size but remaining irregular nanoparticles in shape, indicating that the water fraction of mixed THF/H2O solution had an effect on the morphology of BTE1 to some extent. Maybe, the weeny aggregates were the key of the strong emission of BTE1 in 90% THF/H2O solution.
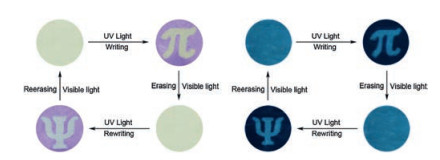
Similar to the THF solution of BTE1, the PMMA film loading with BTE1 aroused a newly broad absorption peak at 560 nm upon the irradiation of 365 nm light (Fig. S20a in Supporting inforamtion). And a clear isosbestic point was observed on 368 nm. These phenomena jointly indicated the photocyclization reaction and the formation of the closed forms could also occur even in PMMA film. The fluorescence of the film also could be quenched upon irradiation of 365 nm light (Fig. S20b in Supporting inforamtion). Besides, the color of the film, the absorption spectra and the emission peak recovered to original state gradually upon irradiation of visible. Those characteristics make BTE1 could be applied in the light controlled soft recording and rewritable printing since the color and the fluorescence of those dyads could reversible switched by light. As shown in Fig. 5, exposed the film to 365 nm UV light for 2 min after covered with a photomask containing a pattern of "π", the letter was recorded on the film whether observed under visible or 300 nm UV light. The film could convert to their original blank-state by erasing the recorded image upon irradiation with visible light for 13 min and applied for the next time of recording. Benefited from the excellent fatigue resistance properties of BTE1, the recording and erasure cycle could perform five times without contrast loss.
To conclude, a series of newly designed unsymmetrical BTEbridge TPE derivatives have been obtained successfully. All those dyads exhibited characteristic photochromism and AIE or AIEE behavior. The results indicated that melding ACQ and AIE units via the BTE-bridge could convert the ACQ-phores to AIE luminophor. The fluorescence of those dyads could be switched ON/OFF upon alternating UV and visible light irradiation both in solution and solid state. The maximum emission wavelength of those dyads could be tuned by different substituent. The PMMA film loading with BTE1 was applied in rewritable information storage successfully. Besides, the electron-withdrawing ability of side substituent could affect the cyclization quantum yields significantly.Aproperly substituent, like aldehyde, could benefit the photoisomerization process. We believed that this work could provide a deeper insight into the rational design of photochromic materials for potential applications in the future. Further development of the AIE characteristic photochromic with various structural modifications, investigation of the structure–activity relationship as well as the incorporation of additional functional moieties to achieve multifunctional photoresponsive materials are now in progress.
This work was financially supported by the National Natural Science Foundation of China (Nos. 21878136, 21372194 and 21773103).
Supplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.040.
M. Irie, T. Fukaminato, K. Matsuda, S. Kobatake, Chem. Rev. 114 (2014) 12174-12277. doi: 10.1021/cr500249p
H. Wu, Y. Chen, Y. Liu, Adv. Mater. 29 (2017) 1605271. doi: 10.1002/adma.201605271
E. Feng, C. Fan, N. Wang, G. Liu, S. Pu, Dyes Pigments 151 (2018) 22-27. doi: 10.1016/j.dyepig.2017.12.041
G. Liu, Y.M. Zhang, L. Zhang, C. Wang, Y. Liu, ACS Appl. Mater. Interfaces 10 (2018) 12135-12140. doi: 10.1021/acsami.7b12822
J. Wang, Y. Gao, J. Zhang, H. Tian, J. Mater. Chem. C 5 (2017) 4571-4577. doi: 10.1039/C7TC00962C
S.C. Pang, H. Hyun, S. Lee, et al., Chem. Commun. (Camb.) 48 (2012) 3745-3747. doi: 10.1039/c2cc30738c
A. Parrot, A. Bernard, A. Jacquart, et al., Angew. Chem. Int. Ed. 56 (2017) 4872-4876. doi: 10.1002/anie.201701860
Q. Qi, C. Li, X. Liu, et al., J. Am. Chem. Soc. 139 (2017) 16036-16039. doi: 10.1021/jacs.7b07738
Q.X. Hua, B. Xin, Z.J. Xiong, et al., Chem. Comm. 53 (2017) 2669-2672. doi: 10.1039/C7CC00044H
M.J. Rust, M. Bates, X. Zhuang, Nat. Methods 3 (2006) 793. doi: 10.1038/nmeth929
E. Betzig, G.H. Patterson, R. Sougrat, et al., Science 313 (2006) 1642. doi: 10.1126/science.1127344
L. Ma, C. Li, Q. Liang, S. Wang, D. Cao, Dyes Pigm. 149 (2018) 543522.
H. Dong, M. Luo, S. Wang, X. Ma, Dyes Pigments 139 (2017) 118-128. doi: 10.1016/j.dyepig.2016.11.054
L. Cui, Y. Baek, S. Lee, N. Kwon, J. Yoon, J. Mater. Chem. C 4 (2016) 2909-2914.
J. Luo, Z. Xie, J.W.Y. Lam, et al., Chem. Comm. (2001) 1740-1741. http://www.ncbi.nlm.nih.gov/pubmed/12240292
Y. Hong, J.W.Y. Lam, B.Z. Tang, Chem. Comm. (2009) 4332-4353. http://www.ncbi.nlm.nih.gov/pubmed/19597589
Z. Ning, Z. Chen, Q. Zhang, et al., Adv. Funct. Mater. 17 (2007) 3799-3807. doi: 10.1002/adfm.200700649
Z. Zhao, J.W.Y. Lam, B.Z. Tang, J. Mater. Chem. 22 (2012) 23726. doi: 10.1039/c2jm31949g
H. Xie, Y. Wu, F. Zeng, J. Chen, S. Wu, Chem. Comm. 53 (2017) 9813-9816. doi: 10.1039/C7CC05313D
D. Li, Y. Zhang, Z. Fan, J. Yu, Chem. Comm. 51 (2015) 13830-13833. doi: 10.1039/C5CC05173H
X. Gu, E. Zhao, T. Zhao, et al., Adv. Mater. 28 (2016) 5064-5071. doi: 10.1002/adma.201505906
J. Wu, S. Sun, X. Feng, et al., Chem. Comm. 50 (2014) 9122-9125. doi: 10.1039/C4CC03127J
J. Mei, N.L.C. Leung, R.T.K. Kwok, J.W.Y. Lam, B.Z. Tang, Chem. Rev. 115 (2015) 11718-11940. doi: 10.1021/acs.chemrev.5b00263
M. Wang, C. Cheng, J. Song, et al., Chin. J. Chem. 36 (2018) 698-707. doi: 10.1002/cjoc.201800115
S.J. Lim, B.K. An, S.D. Jung, M.A. Chung, S.Y. Park, Angew. Chem. Int. Ed. 43 (2004) 6346-6350. doi: 10.1002/anie.200461172
C. Li, H. Yan, G.F. Zhang, et al., Chem. -Asian J. 9 (2014) 104-109. doi: 10.1002/asia.201301071
C. Li, W.L. Gong, Z. Hu, et al., RSC Adv. 3 (2013) 8967-8972. doi: 10.1039/c3ra40674a
S. Chen, W. Li, X. Li, W.H. Zhu, J. Mater. Chem. C 5 (2017) 2717-2722. https://www.researchgate.net/publication/313781347_Aggregation-controlled_photochromism_based_on_a_dithienylethene_derivative_with_aggregation-induced_emission
J. Mei, Y. Hong, J.W.Y. Lam, et al., Adv. Mater. 26 (2014) 5429-5479. doi: 10.1002/adma.201401356
Q. Zhao, Y. Chen, Y. Liu, Chin. Chem. Lett. 29 (2018) 84-86. doi: 10.1016/j.cclet.2017.07.024
F. Wang, X. Li, S. Wang, et al., Chin. Chem. Lett. 27 (2016) 1592-1596. doi: 10.1016/j.cclet.2016.04.020
N.M.W. Wu, H.L. Wong, V.W.W. Yam, Chem. Sci. 8 (2017) 1309-1315. doi: 10.1039/C6SC02928K
Y. Yokoyama, T. Goto, T. Inoue, M. Yokoyama, Y. Kurita, Chem. Lett. 17 (1988) 1049-1052. doi: 10.1246/cl.1988.1049
L. Giordano, T.M. Jovin, M. Irie, E.A. Jares-Erijman, J. Am. Chem. Soc. 124 (2002) 7481-7489. doi: 10.1021/ja016969k

Figure 2 (a) UV–vis spectra changes of BTE1 in THF solutions upon irradiation with 365 nm light depending on different time. Insert: Changes of the photograph of BTE1 in solution upon alternating UV and visible light irradiation. (b) UV–vis spectra of open and closed ring form of BTE1–5 in THF. Concentration: 10-4 mol/L.

Figure 3 (a) Fluorescence spectra of BTE1 in different H2O/THF (v/v) solutions; (b) the dependence of the fluorescence emission intensity on the fw. Insert: Photographs of dyads in different H2O/THF solutions under UV light (300 nm). λex = 300 nm. Concentration: 10-4 mol/L.

Figure 4 (a) Fluorescence spectra change of BTE1 in 90% H2O/THF solution. Insert: Photographs of the solutions upon repeated alternating UV–vis irradiations under 365 nm UV light; (b) Fluorescence spectra change of BTE1 in solid state. Insert: Photographs of the powder upon repeated alternating UV–vis irradiations under 365 nm UV light. λex = 300 nm. Concentration: 10-4 mol/L.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
