
Scheme 1.
Synthetic protocol to synthesize nonsymmetrical aryl thioethers by SNAr reaction of nitroarenes with thiols.

Organic sulfur compounds are essential substances in living organisms and are widely present in biologically active molecules [1, 2]. Thioethers, especially aryl thioethers, are a crucial structural unit in bioactive molecules [3], drugs [4-6] and natural products [7, 8]. The development of synthetic methods for constructing a C–S bond with mild, inexpensive, and easy-to-operate reaction conditions has been attracting long-term, research interests [1, 9-12] since the formation of C–S bonds is one of the most basic reactions in organic synthesis. The traditional method of C–S bond construction is mainly copper-catalyzed Ullmann-type reaction [13, 14]. However, albeit such a reaction typically requires harsh conditions such as high temperature, strong alkali and excess catalyst, the yield is often limited. In the past two decades, continuous efforts of chemists have led to tremendous progresses that with the participation of a catalytic amount of transition metal [15, 16], supplemented by a suitable ligand, C–S bond can be formed under milder conditions. Known catalytic metals mainly include: Cu [17-22], Pd [23-25], Ni [26-29], Co [30], Fe [31-33], Rh [34, 35], Au [36, 37] and Ag [38]. By far, transition metal catalysis is one of the most commonly used and effective means to construct C–S bonds. However, the yield tends to suffer from side reactions due to β-hydride elimination, and more importantly, a costly removal of the transition metal residues is necessary in industrial production. Thus the development of new method for C–S bond construction is highly desired.
An aromatic bimolecular nucleophilic substitution (SN2Ar) reaction typically occurs on aromatic rings with a leaving group under the assistance of one or more electron-withdrawing groups (usually nitro or cyano) in the ortho or para position [39-44] as the activation group. When a nitro group and two or more ortho or para electron-withdrawing groups are present simultaneously on a phenyl ring, the nitro group can also act as a leaving group. However, nucleophilic substitution of a nitrobenzene derivative bearing only one additional electron-withdrawing group is rarely observed [22, 45-48]. In 2006, Oshima and co-workers [49] reported an improved method for SNAr reactions of activated nitroarenes with thiols in dimethylsulfoxide (DMSO) mediated by cesium carbonate. In 2012, Naeimi et al. [50] described another method for SNAr reactions between nitroarenes and thiols in dimethyl formamide (DMF) mediated by magnesium methoxide. These two methods suffer from issues such as the use of a stoichiometric amount of base and high boiling solvents that are inconvenient for product purifications. Our previous research shows that only catalytic amounts of inorganic salts [51] can be used to replace transition metal catalysts. Herein, we report that a catalytic amount of potassium phosphate (K3PO4) can lead to versatile SNAr reactions of activated nitroarenes with thiols in tetrahydrofuran (THF) at room temperature (r.t.) to form an sp2 carbon-sulfur covalent bond (Scheme 1). As far as we know, this is still the first case.
Initially, we screened a series of bases as the potential catalyst with 4-methylphenylthiophenol and para-dinitrobenzene as the model substrates for SNAr reactions of activated nitroarenes with thiols (Table 1, entries 1–9). To our delight, an almost quantitative formation of thioether was observed in THF for 3 h in the presence of a 10 mol% loading of K3PO4 and 20 mol% crown ether (18-crown-6) at room temperature (Table 1, entry 1). Subsequent solvent screening indicated that other solvents, except DMF, led to decreased yields (Table 1, entries 10–14). Further control experiments proved that both the catalyst and the crown ether are necessary for the high yields (Table 1, entries 15 and 16).
With the optimal reaction conditions in hand, we next explored the scope of thiols for the present system (Table 2). Thiophenol derivatives with an either electron-donating or electron-withdrawing group can be efficiently converted to the desired products in good to excellent yields. A wide scope of thiols with various functional groups such as fluoro (entry 10), chloro (entry 11), bromo (entry 12), methoxyl (entry 15), tert-butyl (entry 16), trifluoromethyl (entry 13), ester (entry 8) and heterocycle (entries 17 and 18) are well tolerated. Surprisingly, unprotected amino group is also fairly compatible with the present conditions (entry 14). Aliphatic thiols are also suitable substrates for the present reaction (entries 2–9). It is worth mentioning that a gram-scale version for the reaction between 4-methylphenylthiophenol and para-dinitrobenzene also led to a quantitative yield (entry 1), clearly proving the excellent scalability of the present protocol.
We next studied the scope of nitrobenzene derivatives. Nitrobenzenes bearing moderate to strong electron-withdrawing substituents underwent cross-coupling with thiols to offer the corresponding thioethers. In general, the aryl nucleophilic substitution reaction tend to occur in the ortho or para positions of the electron withdrawing group, but we have found that the meta nitro group can also be substituted to form a thioether in 24% isolated yield (Table 2, entry 1; Table 3, entries 1 and 2). The yield follows the order of para > ortho >> meta. Acetyl nitrobenzenes with different substitution patterns also show the same trend in their reactions with para-methyl thiophenol (Table 3, entries 3–5). The present protocol is also applicable to nitrobenzene derivatives with other strong electron-withdrawing groups, including trifluoromethyl and cyano as well as moderate electron-withdrawing groups, including ester and aldehyde (entries 6–9). However, no reaction was observed for nitrobenzene (entries 10). For substrates with both halo and nitro substituents, selective substitution of halogen, rather than nitro, by the thiol occurred (entries 11–13), indicating that halogens are better leaving groups than nitro under current conditions. Interestingly, for nitropyridine derivatives, both electron-donating and electron-withdrawing groups are suitable substrates (entries 14–17). In sharp contrast to nitrobenzene derivatives, chloro group on the pyridine ring is well tolerated. In addition, the bromine at the 3 position on the pyridine ring is also compatible, but what surprised us was that bromine was preferentially replaced when it was at the 2 position (entry 18).
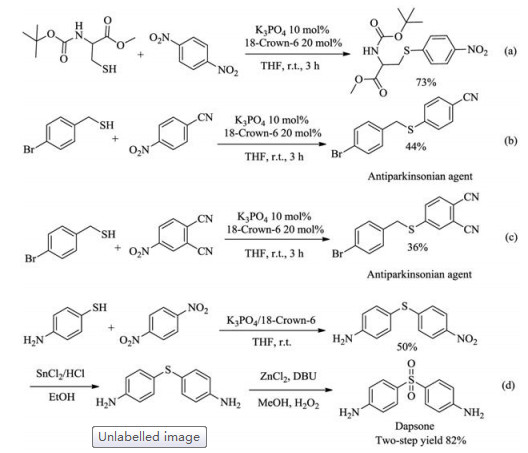
Encouraged by the wide scope of the present protocol, we next evaluated its potential applications in syntheses of biologically relevant compounds. To our delight, protected cysteine can also react with para-dinitrobenzene to form the desired C–S bond (Scheme 2a). Two antiparkinsonian agents [52], 4-((4-bromobenzyl)-thio)benzonitrile and 4-((4-bromobenzyl)thio)phthalonitrile, were also successfully synthesized via the present protocol (Scheme 2b and c). By taking advantages of the classical transformations of nitro group [53], the synthetic utilities of the present method can be further expanded. For example, the reaction of para-aminothiophenol and para-dinitrobenzene via the present protocol allows for an efficient synthesis of Dapsone, a drug for the treatment of leprosy [54], upon reduction of the nitro group in the coupling product to an amino group followed by oxidation of thioether to sulfone (Scheme 2d) [55].
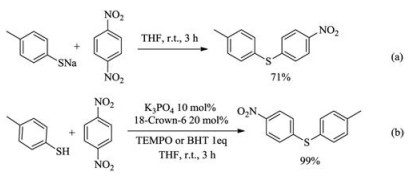
We next performed experiments to gain insights into the reaction mechanism. It was found that sodium para-tolylthiolate reacted with para-dinitrobenzene to afford the coupling product in 71% yield without adding any catalyst (Scheme 3a), which suggests that the salt may just act as a base in the catalysis. The C–S coupling process might operate through either a bimolecular nucleophilic (SN2Ar) substitution pathway or a radical one [9, 56] one due to electron transfer between the thiolate and electron-deficient arenes. It was found that the reaction was not inhibited by the addition of radical trap TEMPO or dibutylhydroxytoluene (BHT) (Scheme 3b), disfavoring the radical pathway. Theoretically, the SNAr reaction has two reaction mechanisms—a stepwise one via Meisenheimer intermediate or a concerted one with any intermediate. We next adapted a literature procedure [57] to investigate the kinetic isotope effect of 12C/13C on the C–S coupling reactions of dinitrobenzene and 1-(4-nitrophenyl)ethan-1-one, affording KIE of 1.014 and 1.011, respectively (details see Supporting information). Based on the correlation between experimentally measured 13C KIE and theoretically identified mechanism of selected 120 SNAr reactions [58], the observed KIE values suggest that the reaction is more in line with the stepwise mechanism. So we believe that one feasible mechanism is that the thiol first forms RS− under the action of a catalyst, and the formed RS− acts as a nucleophile to attack the activated nitrobenzene to form the Meisenheimer complex, and finally removes the nitro group to form the target product (Scheme 4).

In summary, we have developed a convenient catalytic approach for the synthesis of aryl sulfides and diaryl sulfides via potassium phosphate-catalyzed in good to excellent yields by SNAr reactions of activated nitrobenzenes with thiols. The novelty of the methodology, the simplicity of the operation, and the mildness of the reaction conditions make the process an attractive tool to C–S bond formation.
We acknowledged the financial support by the National Natural Science Foundation of China (No. U1532135).
Supplementary material related to this article canbefound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2019.07.012.
K.L. Dunbar, D.H. Scharf, A. Litomska, C. Hertweck, Chem. Rev.117(2017) 5521-5577. doi: 10.1021/acs.chemrev.6b00697
S.A. Vizer, E.S. Sycheva, A.A. Al Quntar, et al., Chem. Rev.115(2015) 1475-1502. doi: 10.1021/cr4001435
S. Parveen, M.O. Khan, S.E. Austin, et al., J. Med. Chem. 48(2005) 8087-8097. doi: 10.1021/jm050819t
H. Liu, X. Jiang, Chem. Asian J. 8(2013) 2546-2563. doi: 10.1002/asia.201300636
A. Gangjee, Y. Zeng, T. Talreja, et al., J. Med. Chem. 50(2007) 3046-3053. doi: 10.1021/jm070165j
E.A. Ilardi, E. Vitaku, J.T. Njardarson, J. Med. Chem. 57(2014) 2832-2842. doi: 10.1021/jm401375q
T. Tatsuta, M. Hosono, H. Rotinsulu, et al., J. Nat. Prod. 80(2017) 499-502. doi: 10.1021/acs.jnatprod.6b01051
T. Nakazawa, J. Xu, T. Nishikawa, et al., J. Nat. Prod. 70(2007) 439-442. doi: 10.1021/np060593c
M. Fontecave, S. Ollagnier-de-Choudens, E. Mulliez, Chem. Rev. 103(2003) 2149-2166. doi: 10.1021/cr020427j
C. Shen, P. Zhang, Q. Sun, S. Bai, T.S. Hor, X. Liu, Chem. Soc. Rev. 44(2015) 291-314. doi: 10.1039/C4CS00239C
Y. Li, Y. Xu, F. Yang, et al., Chin. Chem. Lett. 30(2019) 222-224. doi: 10.1016/j.cclet.2018.09.014
J. Zhao, X. Jiang, Chin. Chem. Lett. 29(2018) 1079-1087. doi: 10.1016/j.cclet.2018.05.026
L.X. Dai, Prog. Chem. 30(2018) 1257-1297.
F. Monnier, M. Taillefer, Angew. Chem. Int. Ed. 48(2009) 6954-6971. doi: 10.1002/anie.200804497
T. Kondo, T.A. Mitsudo, Chem. Rev. 100(2000) 3205-3220. doi: 10.1021/cr9902749
Y. Li, C. Muck-Lichtenfeld, A. Studer, Angew. Chem. Int. Ed. 55(2016) 14435-14438. doi: 10.1002/anie.201608144
C.G. Bates, P. Saejueng, M.Q. Doherty, D. Venkataraman, Org. Lett. 6(2004) 5005-5008. doi: 10.1021/ol0477935
Y. Jiang, Y. Qin, S. Xie, et al., Org. Lett. 11(2009) 5250-5253. doi: 10.1021/ol902186d
F. Ke, Y. Qu, Z. Jiang, et al., Org. Lett. 13(2011) 454-457. doi: 10.1021/ol102784c
G. Xiao, H. Min, Z. Zheng, G. Deng, Y. Liang, Chin. Chem. Lett. 29(2018) 1363-1366. doi: 10.1016/j.cclet.2017.12.013
R. Xu, J.P. Wan, H. Mao, Y. Pan, J. Am. Chem. Soc. 132(2010) 15531-15533. doi: 10.1021/ja107758d
S.S. Bahekar, A.P. Sarkate, V.M. Wadhai, P.S. Wakte, D.B. Shinde, Catal. Commun. 41(2013) 123-125. doi: 10.1016/j.catcom.2013.07.019
M.A. Fernandez-Rodriguez, Q. Shen, J.F. Hartwig, J. Am. Chem. Soc. 128(2006) 2180-2181. doi: 10.1021/ja0580340
Z. Lian, B.N. Bhawal, P. Yu, B. Morandi, Science 356(2017) 1059-1063. doi: 10.1126/science.aam9041
Z. Qiao, H. Liu, X. Xiao, et al., Org. Lett. 15(2013) 2594-2597. doi: 10.1021/ol400618k
Y. Fang, T. Rogge, L. Ackermann, S.Y. Wang, S.J. Ji, Nat. Commun. 9(2018) 2240. doi: 10.1038/s41467-018-04646-2
M.S. Oderinde, M. Frenette, D.W. Robbins, B. Aquila, J.W. Johannes, J. Am. Chem. Soc. 138(2016) 1760-1763. doi: 10.1021/jacs.5b11244
N. Taniguchi, J. Org. Chem. 69(2004) 6904-6906. doi: 10.1021/jo040184q
X.B. Xu, J. Liu, J.J. Zhang, Y.W. Wang, Y. Peng, Org. Lett. 15(2013) 550-553. doi: 10.1021/ol303366u
Y.C. Wong, T.T. Jayanth, C.H. Cheng, Org. Lett. 8(2006) 5613-5616. doi: 10.1021/ol062344l
A. Correa, M. Carril, C. Bolm, Angew. Chem. Int. Ed. 47(2008) 2880-2883. doi: 10.1002/anie.200705668
H. Wang, L. Wang, J. Shang, et al., Chem. Commun. (Camb.) 48(2012) 76-78. doi: 10.1039/C1CC16184A
J.R. Wu, C.H. Lin, C.F. Lee, Chem. Commun. (Camb.) (2009) 4450-4452.
M. Arisawa, T. Suzuki, T. Ishikawa, M. Yamaguchi, J. Am. Chem. Soc.130(2008) 12214-12215. doi: 10.1021/ja8049996
S.D. Timpa, C.J. Pell, O.V. Ozerov, J. Am. Chem. Soc. 136(2014) 14772-14779. doi: 10.1021/ja505576g
M. Jean, J. Renault, P. van de Weghe, N. Asao, Tetrahedron Lett. 51(2010) 378-381. doi: 10.1016/j.tetlet.2009.11.025
N. Morita, N. Krause, Angew. Chem. Int. Ed. 45(2006) 1897-1899. doi: 10.1002/anie.200503846
R. Das, D. Chakraborty, Tetrahedron Lett. 53(2012) 7023-7027. doi: 10.1016/j.tetlet.2012.09.127
J.F. Bunnett, R.E. Zahler, Chem. Rev. 49(1951) 273-412. doi: 10.1021/cr60153a002
S.D. Ross, M. Finkelstein, J. Am. Chem. Soc. 85(1963) 2603-2607. doi: 10.1021/ja00900a018
F. Pietra, F. Del Cima, J. Org. Chem. 33(1968) 1411-1416. doi: 10.1021/jo01268a021
J.F. Bunnett, R.J. Morath, J. Am. Chem. Soc. 77(1955) 5051-5055. doi: 10.1021/ja01624a033
J.R. Beck, Tetrahedron 34(1978) 2057-2068. doi: 10.1016/0040-4020(78)89004-8
X. Zhang, G.P. Lu, C. Cai, Green Chem. 18(2016) 5580-5585. doi: 10.1039/C6GC01742H
N. Kornblum, L. Cheng, R.C. Kerber, et al., J. Org. Chem. 41(1976) 1560-1564. doi: 10.1021/jo00871a016
J.B. Baumann, J. Org. Chem. 36(1971) 396-398. doi: 10.1021/jo00802a007
A. Rostami, A. Rostami, A. Ghaderi, J. Org. Chem. 80(2015) 8694-8704. doi: 10.1021/acs.joc.5b01248
F.M. Moghaddam, R. Pourkaveh, Catal. Commun. 94(2017) 33-37. doi: 10.1016/j.catcom.2017.02.009
A. Kondoh, H. Yorimitsu, K. Oshima, Tetrahedron 62(2006) 2357-2360. doi: 10.1016/j.tet.2005.11.073
H. Naeimi, M. Moradian, Synlett 23(2012) 2223-2226. doi: 10.1055/s-0032-1317079
C. Fang, C. Lu, M. Liu, et al., ACS Catal. 6(2016) 7876-7881. doi: 10.1021/acscatal.6b01856
M.M. Van der Walt, G. Terre' Blanche, A.C. Lourens, A. Petzer, J.P. Petzer, Bioorg. Med. Chem. Lett. 22(2012) 7367-7370. doi: 10.1016/j.bmcl.2012.10.070
N. Ono, The Nitro Group in Organic Synthesis, John Wiley & Sons, New York, 2001.
Y.I. Zhu, M.J. Stiller, J. Am. Acad. Dermatol. 45(2001) 420-434. doi: 10.1067/mjd.2001.114733
J.B. Feng, J.L. Gong, X.F. Wu, RSC Adv. 4(2014) 29273-29275. doi: 10.1039/C4RA04319G
F. Denes, M. Pichowicz, G. Povie, P. Renaud, Chem. Rev.114(2014) 2587-2693. doi: 10.1021/cr400441m
D.A. Singleton, A.A. Thomas, J. Am. Chem. Soc. 117(1995) 9357-9358. doi: 10.1021/ja00141a030
E.E. Kwan, Y. Zeng, H.A. Besser, E.N. Jacobsen, Nat. Chem. 10(2018) 917-923. doi: 10.1038/s41557-018-0079-7

Scheme 1 Synthetic protocol to synthesize nonsymmetrical aryl thioethers by SNAr reaction of nitroarenes with thiols.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:



 下载:
下载:


