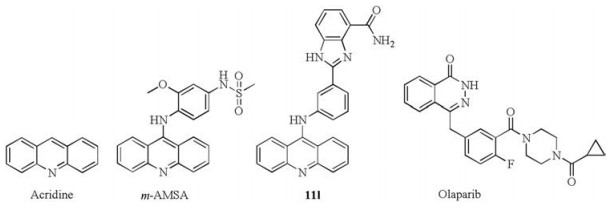
Figure 1.
Acridines and phthalazinone derivatives.
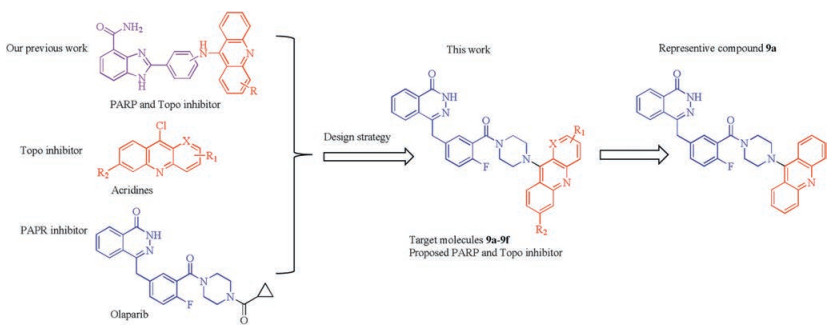
Design, synthesis and biological evaluation of novel phthalazinone acridine derivatives as dual PARP and Topo inhibitors for potential anticancer agents
Qiuzi Dai , Jiwei Chen , Chunmei Gao , Sun Qinsheng , Zigao Yuan , Jiang Yuyang

The structural integrity and stability of DNA are important for cell survival and normal physiological functions [1-3]. However, some endogenous or exogenous factors could directly or indirectly induce DNA damages or even mutation [4-6], which, if unrepaired, would lead to the development of various diseases, including cancer. Targeting pathways of DNA damage and repair are feasible strategy for treatment of cancer [7-9].
Topoisomerases (Topos) are necessary for the genome DNA correctly squeezed in a small cell nuclear. According to their structure and catalytic mechanism, Topos are classified into two major classes, during which Topo Ⅰ cleaves one single-stranded DNA during each catalytic cycle, while Topo Ⅱ cleaves double stranded DNA to resolve the DNA damage problems [10, 11]. Overexpression of Topos may cause the instability of genome DNA and have been observed in many tumor cells. Targeting Topos have been demonstrated to be an effective way for cancer therapy [12-14]. Many acridine analogues, such as m-AMSA (Fig. 1), have entered clinical or preclinical trials as potent Topo inhibitors. Our group has also devoted much efforts in developing acridine-based Topo inhibitors for cancer therapy [15-19].
At the same time, poly(ADP-ribose) polymerases (PARPs) are well-known sensors of DNA damage and could repair the singlestrand breaks (SSBs) in DNA via the base-excision repair (BER) pathway [20-22]. PARP-1 is a well-established target for developing anti-tumor drugs. PARP inhibitors could block PARP-mediated DNA damage repair and make tumor cells more sensitivity to cytotoxic agents. During the last decades, many PARP inhibitors (PARPi) have been reported [23-25]. Olaparib (Fig. 1) is the first PARP inhibitor approved by the FDA in 2014 for the treatment of advanced ovarian cancer. Different combinations of PARP inhibitors and genotoxic drugs are proceeded in clinical trials. These combinations including alkylating agents (temozolomide), crosslinking agents (cisplatin), Topo Ⅰ inhibitors (topotecan and irinotecan) and Topo Ⅱ inhibitors (etoposide) [26].
In this study, we intended to develop dual PARP and Topo inhibitors based on rational drug design strategy and our own studies reported previously [15, 27-31]. We have developed 4-amidobenzimidazole acridine derivatives as first-in-class dual PARP and Topo inhibitors for cancer therapy [17]. As reported, compound 11 l (Fig. 1) showed good in vitro activities targeting Topo and PARP-1 and could efficiently suppress tumor growth in mice. Considering the relationship between PARP and Topo and the significant results, we hold the opinion that simultaneously targeting Topo and PARP-1 is a promising strategy for combating cancer. Phthalazinones has been recognized as an effective scaffold to develop PARP inhibitors especially since the approval of olaparib [32-34]. The phthalazinone structure of olaparib is a vital functional group to interact with PARP and the piperazine moiety is mainly used to improve the activity or adjust the physical and chemical properties. Therefore, the piperazine moiety can be modified or optimized without loss of PARP inhibitory activity based on the retention of the phthalazinone functional group (Scheme 1). Herein we developed a series of phthalazinone acridine derivatives by changing the substituent pattern of the acridine group and further evaluated their anti-tumor activities.
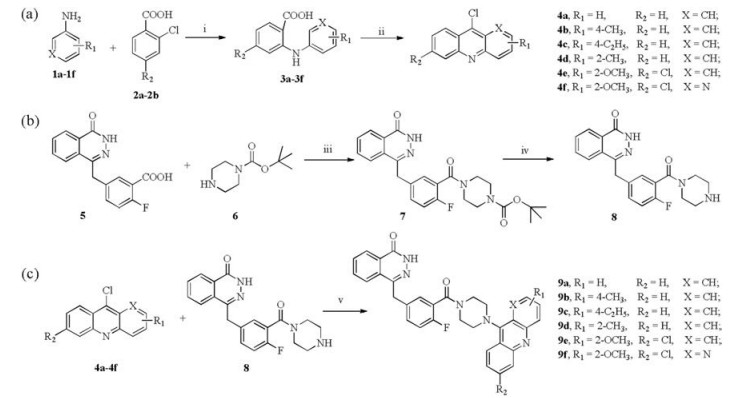
The synthesis of target molecules 9a-9f is shown in Scheme 2. Firstly, compounds 3a-3f were obtained by using 2-chlorobenzoic acid 2a-2b reacted with phenylamine derivatives 1a-1f via the Ullmann reactions. Then Friedel-Crafts acylation reactions were conducted in phosphorus oxychloride at 105 ℃ for 4 h to yield the corresponding acridine intermediates 4a-4f [35]. Secondly, Compound 8 were synthesized according to the reported procedures [36]. At last, the desired compounds 9a-9f were produced by the nucleophilic substitution reactions between the phthalazinone derivative 8 and corresponding 9-chloroacridines 4a-4f. Target compounds were characterized with 1H NMR, 13C NMR, melting point and high resolution mass spectrum (Supporting information).
According to our previous studies, the size, length and the electron-negativity of the substituents on the acridine ring and the linker between two kinds of inhibitors had great effect on the antitumor activity [28]. Besides, methyl and methoxyl substitution at 2-position and 4-position of acridine could be helpful to its bioactivity as dual inhibitors of PARP and Topo [17]. In this report, PARP inhibitor of phthalazinone conjugated with Topo inhibitor acridine was used as a new scaffold compound for the development of PARP and Topo dual inhibitors. Compounds that were substituted by methyl, methoxyl or chloride substituted on the acridine ring were investigated. Using m-AMSA and olaparib as positive control, the cellular activities of the compounds were evaluated by the MTT assays. Different kinds of cancer cells including MCF-7 (human breast adenocarcinaoma cell line), K562 (human chronic myelogenous leukemia cells), HepG2 (human hepatoma cells), HeLa (human cervical cancer cells), HCT116 (human colon cancer cell line) and HCC1937 (human breast cancer cell line) were tested. Among the compounds we synthesized (Table 1), 9a and 9c displayed outstanding antiproliferative activities in all the tumor cells mentioned above. In particular, 9a and 9c showed similar activities to olaparib in MCF-7 and HCC1937 cells and greatly improved activities in the others. Further, compared with 9c, 9a with non-substitution of acridine showed better activities. While, compound 9f with the N substituted at 1-position of acridine exhibited significantly reduced activity. In terms of cellular activity, 9a is the most potent one among the compounds.
 DownLoad:
CSV
DownLoad:
CSV

|
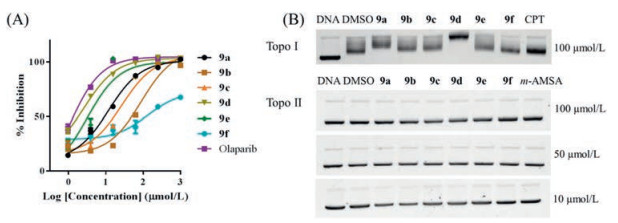
We then tested the activities of the synthesized compounds targeting PARP1 and Topo Ⅰ/Ⅱ enzymes using olaparib, camptothecin (CPT) and m-AMSA as positive controls. As shown in Fig. 2A and Table 1, compounds 9a and 9c showed slightly weak PARP-1 inhibitory activities compared to olaparib. Compounds 9d and 9e showed similar PARP-1 inhibitory activities compared to olaparib. Compounds 9b and 9f showed poor PARP-1 inhibitory activities compared to olaparib. Compound 9d showed the most potent PARP-1 inhibitory activity among the synthesized compounds, while its antiproliferative activities against cancer cells were weak (Table 1), which may attribute to the compound's physicochemical properties (e.g., solubility, permeability, lipophilicity, and stability) or the cellular environments (e.g., cell membrane, pH, and metabolism) which could attenuate the amounts of compounds reaching the therapeutic target. The weak activity of 9f against tumor cells and PARP1 might result from unfavorable solubility (Fig. S1 and Table S1 in Supporting information). Moreover, all the compounds displayed comparable Topo Ⅱ inhibitory activities to m-AMSA at 10 μmol/L (Fig. 2B). These data suggest that all the compounds could potently inhibit Topo Ⅱ, warranting them as dual PARP and Topo inhibitors.
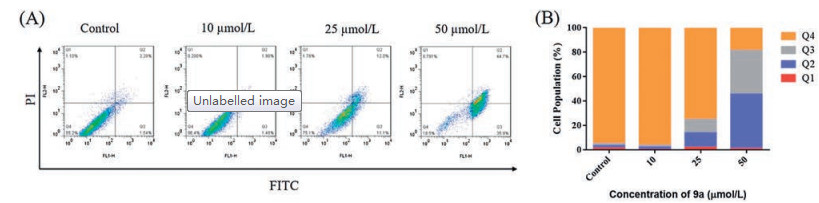
DNA damage and its repair have been reported to have effect on the apoptosis pathway [37]. We choose 9a for further study as it showed excellent cellular activities and desirable enzyme activities. To evaluate whether compound 9a could induce apoptosis, an annexin-V/PI binding assay was conducted in HCT116 cells as shown in Fig. 3. Compared with DMSO controls, HCT116 cells treated with 9a at different concentrations of 10, 25, and 50 μmol/L resulted in 0.5%, 22.9%, and 81.5% cells apoptosis, respectively, suggesting that compound 9a could induce apoptosis in a dose dependent manner.
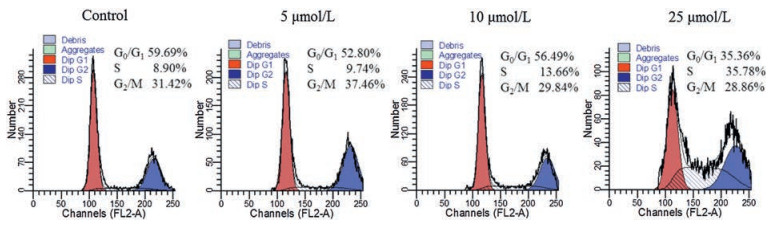
DNA damage caused by antitumor agents may activate DNA damage checkpoints, which arrest cell cycle progression, initiate DNA damage repair, or lead to cell death [37]. To evaluate whether compound 9a could induce tumor cell cycle arrest, we then performed a flow cytometry assay to assess the effect of compound 9a on the distribution of cell cycle. The cell cycle profiles of HCT116 after treating with compound 9a in different concentrations for 48 h were illustrated in Fig. 4. The S-phase fraction was gradually increased from 8.90% in the untreated cells to 9.74%, 13.66%, and 35.78% in cells treated with compound 9a at 5, 10, and 25 μmol/L, respectively, indicating that compound 9a caused S cell cycle arrest in a dose-dependent manner.
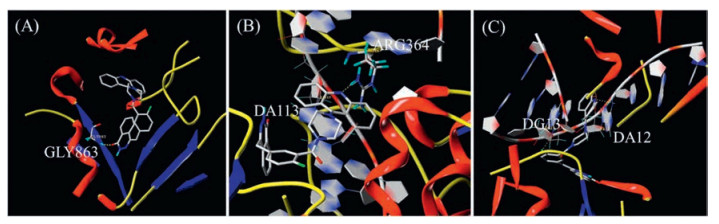
To further understand the mechanism of compound 9a interacting with the respective enzymes, 9a was docked into the active sites of PARP1 (PDB code: 4UND), Topo Ⅰ (PDB code: 1 K4T) and Topo Ⅱ (PDB code: 4 G0U) using the SYBYL-X 1.3 protocol. As shown in Fig. 5A, the phthalazinone group of 9a occupied the nicotinamideribose binding domain of PARP1, forming a hydrogen bond with Gly863. Moreover, the nitrogen atom of the acridine group formed one more hydrogen bond with Arg364 of Topo Ⅰ and the π-π stacking interaction between acridine with DA113 was observed (Fig. 5B). The docking results suggested that 9a could form cleavage-complex with Topo Ⅰ and DNA. Besides, the phthalazinone group of 9a was parallel to DA12 and formed two hydrogen bonds with DA12 and DG13, leading to the formation of cleavage-complex with Topo Ⅱ and double strands DNA (Fig. 5C). The above docking results may further confirm that 9a could act as dual PARP and Topo Ⅰ/Ⅱ inhibitors.
In conclusion, Topos and PARPs are important for genome stability. Inhibitors targeting both of the targets may have synergistic antitumor effects. In this study, we designed and synthesized a new series of phthalazinone acridine derivatives targeting both PARP and Topo. Some of the compounds displayed potent antiproliferative activities against kinds of cancer cells and showed potential inhibitory potency against both PARP-1 and Topo Ⅰ/Ⅱ in vitro. Compound 9a exhibited the most potent antiproliferative activity in its class. Further evaluation indicated that 9a induced remarkable apoptosis, and caused prominent S cell cycle arrest. Taken together, compound 9a is a promising dualtargeted inhibitor and is worth for further optimization for cancer therapy.
The authors would like to thank the financial supports from the Shenzhen Development and Reform Committee (Nos. 20151961 and 2019156) and Department of Science and Technology of Guangdong Province (No. 2017B030314083).
Supplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.019.
A.J. Deans, S.C. West, Nat. Rev. Cancer 11(2011) 467-480. doi: 10.1038/nrc3088
T. Helleday, E. Petermann, R.A. Sharma, et al., Nat. Rev. Cancer 8(2008) 193-204. doi: 10.1038/nrc2342
M.J. O'Connor, Mol. Cell. 60(2015) 547-560. doi: 10.1016/j.molcel.2015.10.040
A. Athie, S.J. Carles, J. Mateo, et al., Cancer Curr. Oncol. Rep. 21(2019) 42-42. doi: 10.1007/s11912-019-0790-6
M. de Vos, V. Schreiber, F. Dantzer, Biochem. Pharmacol. 84(2012) 137-146. doi: 10.1016/j.bcp.2012.03.018
N. Hustedt, D. Durocher, Nat. Cell Biol. 19(2017) 1-9. doi: 10.1038/ncb3452
J.S. Brown, B. O'Carrigan, S.P. Jackson, T.A. Yap, Cancer. Discov. 7(2017) 20-37. doi: 10.1158/2159-8290.CD-16-0860
A.R. Chaudhuri, A. Nussenzweig, Nat. Rev. Mol. Cell. Biol. 18(2017) 610-621. doi: 10.1038/nrm.2017.53
P.A. Jeggo, L.H. Pearl, A.M. Carr, Nat. Rev. Cancer 16(2016) 35-42. doi: 10.1038/nrc.2015.4
I.D. Connolly, J.D. Hixson, S. Nagpal, Drug. Future 41(2016) 731-740. doi: 10.1358/dof.2016.041.12.2563475
D.C. Hooper, G.A. Jacoby, C.S.H. Perspect. Med. 6(2016) 5320-5342. doi: 10.1101/cshperspect.a025320
A. Canela, R. Casellas, A. Nussenzweig, et al., Cell 170(2017) 507-521. doi: 10.1016/j.cell.2017.06.034
S.M. Cuya, M.A. Bjornsti, R.C.A.M. van Waardenburg, Cancer Chemother. Pharm. 80(2017) 1-14. doi: 10.1007/s00280-017-3334-5
X. Liang, Q. Wu, W. Zhang, et al., Eur. J. Med. Chem. 171(2019) 129-168. doi: 10.1016/j.ejmech.2019.03.034
J. Chen, D. Li, Y. Jiang, et al., Bioorg. Med. Chem. 26(2018) 3958-3966. doi: 10.1016/j.bmc.2018.06.016
Z. Cui, S. Chen, Y. Jiang, et al., Eur. J. Med. Chem. 136(2017) 372-381. doi: 10.1016/j.ejmech.2017.05.006
Z. Yuan, C. Gao, Y. Jiang, et al., Eur. J. Med. Chem. 138(2017) 1135-1146. doi: 10.1016/j.ejmech.2017.07.050
B. Zhang, F. Liu, Y. Jiang, et al., Eur. J. Med. Chem. 129(2017) 337-348. doi: 10.1016/j.ejmech.2017.02.027
W. Zhang, H. Liu, Y. Jiang, et al., Eur. J. Med. Chem. 116(2016) 59-70. doi: 10.1016/j.ejmech.2016.03.066
R.J. Henning, M. Bourgeois, R.D. Harbison, Cardiovasc. Toxicol. 18(2018) 493-506. doi: 10.1007/s12012-018-9462-2
P.G. Jain, B.D. Patel, Eur. J. Med. Chem. 165(2019) 198-215. doi: 10.1016/j.ejmech.2019.01.024
K.N. Moore, M.R. Mirza, U.A. Matulonis, Gynecol. Oncol. 149(2018) 214-220. doi: 10.1016/j.ygyno.2018.01.011
R. Gupte, Z. Liu, W.L. Kraus, Genes Dev. 31(2017) 101-126. doi: 10.1101/gad.291518.116
L. Livraghi, J.E. Garber, BMC Med. 13(2015) 188-204. doi: 10.1186/s12916-015-0425-1
C.J. Lord, A. Ashworth, Science 355(2017) 1152-1158. doi: 10.1126/science.aam7344
J. Murai, J.H. Doroshow, Y. Pommier, et al., J. Pharmacol. Exp. Ther. 349(2014) 408-416. doi: 10.1124/jpet.113.210146
C. Ding, C.Y. Tan, Y.Y. Jiang, et al., Chin. Chem. Lett. 28(2017) 1220-1227. doi: 10.1016/j.cclet.2017.01.003
C. Gao, F. Liu, Y. Jiang, et al., Bioorg. Med. Chem. 23(2015) 1800-1805. doi: 10.1016/j.bmc.2015.02.036
Z. Yuan, Y. Chen, Y. Jiang, et al., Bioorg. Chem. 87(2019) 200-208. doi: 10.1016/j.bioorg.2019.03.027
Z. Yuan, C. Tan, Y. Jiang, et al., Bioorg. Med. Chem. 25(2017) 4100-4109. doi: 10.1016/j.bmc.2017.05.058
Z. Yuan, C. Tan, Y. Jiang, et al., Eur. J. Med. Chem. 134(2017) 281-292. doi: 10.1016/j.ejmech.2017.04.017
H. Almahli, N.S. Al-shakliah, W.M. Eldehna, et al., Bioorg. Chem. 77(2018) 443-456. doi: 10.1016/j.bioorg.2018.01.034
K.A. Menear, G.C.M. Smith, N.M.B. Martin, et al., Eur. J. Med. Chem. 97(2015) 462-482. doi: 10.1016/j.ejmech.2014.11.043
L.X. Wang, X.B. Zhou, S. Li, et al., Bioorg. Med. Chem. Lett. 24(2014) 3739-3743. doi: 10.1016/j.bmcl.2014.07.001
C. Gao, F. Liu, Y. Jiang, et al., Bioorg. Chem. 60(2015) 30-36. doi: 10.1016/j.bioorg.2015.04.002
F. Zmuda, A. Sutherland, S.L. Pimlott, et al., J. Med. Chem. 58(2015) 8683-8686. doi: 10.1021/acs.jmedchem.5b01324
A. Skladanowski, P. Bozko, M. Sabisz, Chem. Rev. 109(2009) 2951-2973. doi: 10.1021/cr900026u

Scheme 2 Synthesis of compounds 9a-9f. Reagent and conditions: (a) () Cu, K2CO3, DMF, 130 ℃, 4 h; (ⅱ) POCl3, 105 ℃, 2 h; (b) (ⅲ) HATU, trithylamine, DMF, 2 h, room temperature; (ⅳ) HCl, H2O, EtOH, 3 h, room temperature; NH3·H2O, CH2Cl2; (ⅴ) Phenol, Ar, 120 ℃, 1 h.

Figure 2 PARP-1, Topo Ⅰ and Topo Ⅱ inhibitory activities. (A) PARP-1 inhibition activities of compounds 9a-9f and olaparib at different concentrations. The IC50 values were determined by fitting the experiment data to log(inhibitor) vs. response-variable slope (three parameters) model in Prism. (B) Topo Ⅰ: lane DNA: pBR322 DNA; lane DMSO: Topo Ⅰ + pBR322 DNA; lane CPT: camptothecin + Topo Ⅰ + pBR322 DNA; the others: compounds (100 μmol/L) + Topo Ⅰ + pBR322 DNA. Topo Ⅱ: lane DNA: pBR322 DNA; lane DMSO: Topo Ⅱ + pBR322 DNA; lane m-AMSM: m-AMSM + Topo Ⅱ + pBR322 DNA; the others: tested compounds at defined concentrations + Topo Ⅱ + pBR322 DNA.

Figure 3 Compound 9a induced apoptosis. (A) HCT116 cells were incubated with different concentrations of compounds 9a for 24 h followed by annexin V and PI staining and analyzed by flow cytometry. (B) Apoptosis analyzed by cell population distributions.

Figure 5 Molecular models of Compound 9a docked into (A) PARP-1, (B) Topo Ⅰ & DNA complex and (C) Topo Ⅱ & DNA complex.
Table 1. In vitro antiproliferative potency and PARP-1 inhibitory activity of compounds 9a–9f.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


