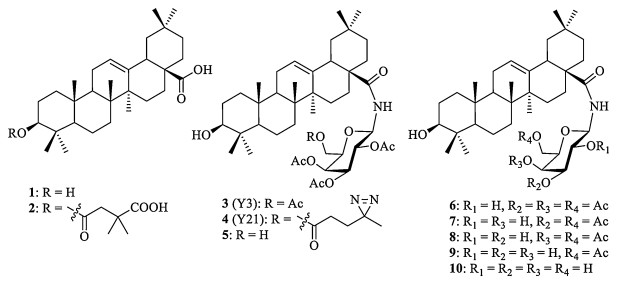
Figure 1.
Chemical structures of oleanolic acid (OA, 1) and its derivatives (2-10).
Selective and facile deacetylation of pentacyclic triterpenoid under methanolic ammonia condition and unambiguous NMR analysis
Han Wang , Renyang Xu , Shuobin Liang , Fuxiang Ran , Lihe Zhang , Yongmin Zhang , Demin Zhou , Sulong Xiao

Oleanolic acid (OA, 1) is a naturally occurring oleanane-type pentacyclic triterpene (Fig. 1). It is widely distributed in the plant kingdom in more than 1, 620 plant species [1]. The compound is especially prevalent in plants belonging to the Oleaceae family, such as the bitter olive Olea europaea (leaves, fruit) [2], and it still serves as the main source of commercial OA [3]. OA and its derivatives display several interesting biological and pharmacological activities, including anticancer, anti-inflammatory and antioxidant properties [4, 5]. Due to its low toxicity and excellent activity, the antiviral properties of OA have attracted increasing attention in recent years. Kashiwada et al. have found that OA possesses anti-HIV activity with an EC50 value of 3.7 μmol/L and it can inhibit the growth of uninfected H9 cells with an IC50 value of 47.8 cmol/L [6]. As a result of structural modification of OA, the introduction of a 3, 3-dimethylsuccinyl group at C3OH generates the most potent compound 2, which has been shown to have antiHIV-1 activity with an EC50 value of < 0.0005 μg/mL and a therapeutic index of 22, 400 [6].
In our recent study, we have found that Y3 (3), an o leanane-type triterpenoid saponin, exhibits significant inhibitory effect on influenza virus entry by tightly binding to the pocket within viral envelope hemagglutinin (HA) for sialic acid receptor. Docking studies suggest that the amide of peptide bond A137 of HA, a critical residue for hydrogen bonding to the carboxylate of sialic acid, forms a hydrogen bond with the 2-estercarbonyl group of the galactose of 3, which is also hydrogen bonded to the amide of residue Q226. Another hydrogen bond is formed between S145 of HA1 and the 3-carbonyl group of the galactose of 3 (Fig. S1 in Supporting information) [7]. Diazirine-based photoaffinity labeling has been proven to be a powerful tool to probe proteins by attaching a diazirine group to the corresponding small molecule ligands [8-11]. To directly identify the target site of inhibitor 3, photo-reactive diazirine group is selected for labeling pentacyclic triterpenoid. Y21 (4), a photoaffinity probe containing diazirine group at the C6'-OH of galactose, is further designed and synthesized [12]. The incubation of 4 with HA protein followed by MS analysis of the UV-activated mixture reveals that heptad repeat-2 (HR2), the prevalent heptad repeat sequence comprising an α-helical coil in viral fusion proteins, is the direct target in the triterpenoid-mediated inhibition of influenza virus fusion.
During the synthesis of the photocrosslinking probe 4, we have attempted to obtain its precursor 5 from 3 by simple selective deacetylation at C6 of the galactose. In 2000, Orita et al. have reported that the primary acetate can be selectively removed in multitude of carbohydrate polyacetates with the neutral organotin catalyst [tBu2SnOH(Cl)]2 [13, 14]. As the procedure requires expensive reagents and catalysts, methanolic ammonia condition was selected for the deacetylation reaction in our work. To our surprise, good selectivity for cleaving gal-C2-OAc group of 3 was achieved within 4 h at low temperature (-60℃) in a yield of 56%. This approach was simpler than the conventional procedure that usually requires many steps, including temporary regioselective protection and deprotection. When the reaction temperature was increased from -60℃ to room temperature, four other deacetylation products were obtained with reasonable yields. In the present study, we would like to describe the synthesis and unambiguous structure characterization of the five deacetylated products 6-10 from 3 under methanolic ammonia condition.
As shown in Scheme 1, we submitted triterpenoid 3 to the action of liquid ammonia in CH3OH/THF (1:1, v/v) solvents and observed that the deacetylation reactions were closely dependent on the reaction conditions, especially the temperature (Scheme 1).
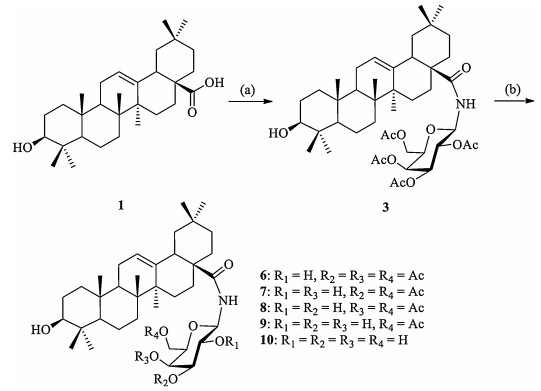
Four sets of conditions were employed to access the deprotection of acetyl groups. Table 1 summarized the yields of the products and reaction conditions. Apparently, the reaction temperature and time were significant factors in discriminating the protecting groups. When triterpenoid 3 was treated with methanolic ammonia under nitrogen for 12 h at -60℃ (entry 1), only one compound 6, with the acetyl group at gal-C2 deprotected, was obtained in a yield of 56%. Unlike the reported organotin catalyst [14], the acetyl group from primary alcohol was deprotected. Further treatment of 3 under the same conditions but a higher reaction temperature (-40℃) (entry 2) gave compound 6 as the major product (~50%) as expected, along with two minor 2, 4-and 2, 3-double deacetylation products 7 and 8 (less than 10%), respectively. Therefore, a prolonged reaction time was required for satisfactory yields of 7 and 8 (12 h, 8% and 23% yields, respectively). We observed that 8 was more polarized than its isomer 7 (on the basis of its migratory ability on silica gel). The two compounds should be yielded from compound 6 after further deacetylation at gal-C3 or gal-C4. The yield of 8 was almost three times of 7 (23% vs. 8%), suggesting that the acetyl group at gal-C3 was more easily to be deprotected than that at gal-C4. If the reaction mixture was warmed-up from -40℃ to -20℃ for 4 h (entry 3), the reaction led to the formation of four products 6-9 with yields ranging from 17% to 24%. Notably, further warm-up of the reaction mixture from -20℃ to ambient temperature (25℃) (entry 4) resulted in a quantitative yield of 10, with four acetyl groups at galactose removed completely, within 2 h.
 DownLoad:
CSV
DownLoad:
CSV

|
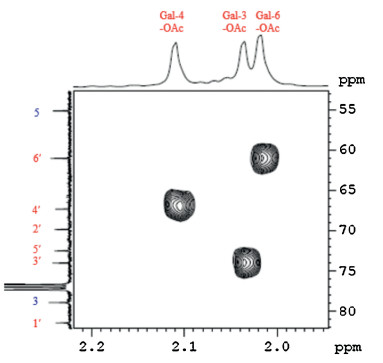
The structural determination of compounds 6-9 were achieved using one and two dimensional NMR spectroscopy and ESI-HRMS, which are provided in the Supporting Information. The key to the structure characterization of these compounds was to determine the acetyl groups' position. Based on the 1H-1H COSY and 1H-13C HSQC spectra, the 1H and 13C signals of galactose moiety were determined easily (Tables S1 and S2 in Supporting information). Then 1H-13C HMBC spectrum was used to determine the position of acetyl groups. In a typical example, the 1H and 13C signals of galactose moiety of compound 6 were unambiguously assigned with the aid of 1H-13C HSQC (Fig. S2 in Supporting information). The HMBC spectrum of 6 further disclosed long-range 1H-13C correlations of the high-field signals at dH 2.11, 2.04 and 2.02, which were assigned to three acetyl groups, with gal-C4 (δC 67.32), gal-C3 (δC 74.01) and gal-C6 (δC 61.03) (Fig. 2), respectively. These finding indicated that the deacetylation reaction occurred at gal-C2 position of 3. Similarly, the assignment of the acetyl groups of compounds 7-9 was also carried out based on the HMBC spectra (Figs. S3-S5 in Supporting information). Compound 10 was identified as the known compound, and the 1H and 13C NMR spectra were consistent with previously reported data [7].
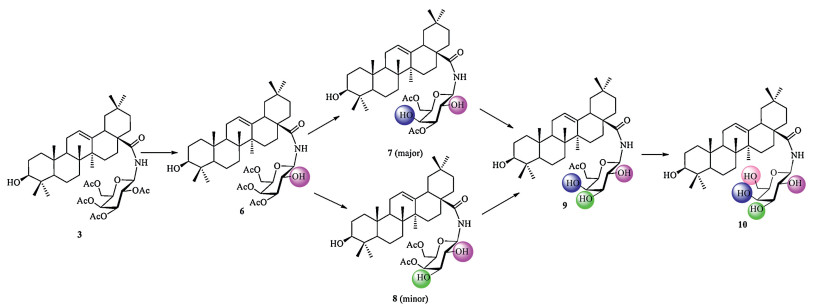
Based on these results and the structures of compounds 6-10, we would like to propose a plausible route for the deacetylation reaction of compound 3 under methanolic ammonia (Fig. 3). In the first step, the acetyl group at gal-C2 position was selectively deprotected, yielding compound 6. Then the acetyl group at gal-C4 or gal-C3 was removed to transform 6 into 7 or 8 simultaneously. Further deacetylation gave compound 9. Finally, the acetyl group at gal-C6 was removed to give compound 10.
We next investigated the applicability of the protocol used above for per-O-acetylated galactose. Except the acetyl group at gal-C1, similar deacetylation reactions were also found for the other acetyl groups and the ESI-HRMS spectra of the partial deacetylation products were provided in the Supporting information (Figs. S6-S9 in Supporting information). The use of this method for the deacetylation of other pentacyclic triterpenoids as well as carbohydrates, including monosaccharides, disaccharides, etc., are currently carried out in our laboratory.
In summary, methanolic ammonia was proved to be an efficient reagent for the deacetylation of pentacyclic triterpenoid. This protocol provided a rapid access to the diversified triterpenoid derivatives, which frequently require orthogonal protection during the course of synthesis. By this effective approach, C2-OAc group of galactose was first cleaved at low temperature with good selectivity, and a series of partially deacetylated OA-galactose derivatives were separated and unambiguously characterized. Further applications of this method and biological evaluation of these new compounds are currently under investigation.
This work was supported by the National Natural Science Foundation of China (Nos. 21572015, 21877007, 81703540 and 21702007), China Postdoctoral Science Foundation (No. 2018M631796), Technology Plan Foundation of Liaoning Province (No. 20170520063), Chinese Medicine Related Scientific Research Project of Dalian (No.17Z2013) and the open funding of the State Key Laboratory of Phytochemistry and Plant Resources in West China.
Supplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.007.
J. Pollier, A. Goossens, Phytochemistry 77(2012) 10-15. doi: 10.1016/j.phytochem.2011.12.022
I. Kubo, A. Matsumoto, Experientia 40(1984) 937-938. doi: 10.1007/BF01946446
M.B. Sporn, K.T. Liby, M.M. Yore, et al., J. Nat. Prod. 74(2011) 537-545. doi: 10.1021/np100826q
C. Lin, X.A. Wen, H.B. Sun, Expert Opin. Ther. Pat. 26(2016) 643-655. doi: 10.1080/13543776.2016.1182988
K. Xu, F.H. Chu, G.L. Li, et al., Pharmazie 69(2014) 483-495. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM25073392
Y. Kashiwada, H.K. Wang, T. Nagao, et al., J. Nat. Prod. 61(1998) 1090-1095. doi: 10.1021/np9800710
M. Yu, L. Si, Y. Wang, et al., J. Med. Chem. 57(2014) 10058-10071. doi: 10.1021/jm5014067
M. Hashimoto, Y. Hatanaka, Eur. J. Org. Chem. 2008(2008) 2513-2523. doi: 10.1002/ejoc.200701069
L. Dubinsky, B.P. Krom, M.M. Meijler, Bioorg. Med. Chem. 20(2012) 554-570. doi: 10.1016/j.bmc.2011.06.066
M.W. Halloran, J.P. Lumb, Chem. -Eur. J. 25(2019) 4885-4898. doi: 10.1002/chem.201805004
J.R. Hill, A.A.B. Robertson, J. Med. Chem. 61(2018) 6945-6963. doi: 10.1021/acs.jmedchem.7b01561
L.L. Si, K. Meng, Z.Y. Tian, et al., Sci. Adv. 4(2018) eaau8408. doi: 10.1126/sciadv.aau8408
A. Orita, K. Sakamoto, Y. Hamada, et al., Synlett 2000(2000) 140-142. doi: 10.1055/s-2000-6474
A. Orita, Y. Hamada, T. Nakano, et al., Chem. -Eur. J. 7(2001) 3321-3327. doi: 10.1002/1521-3765(20010803)7:15<3321::AID-CHEM3321>3.0.CO;2-H

Scheme 1 Reagents and conditions: (a) 2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosylamine, EDC, THF, 62%; (b) NH3 (L), CH3OH/THF (1:1, v/v), -60℃ to r.t.

Figure 2 A portion of the 400 MHz HMBC (CDCl3, 25℃) spectrum of 6, with the 1D 1H and 13C NMR spectra along the top and the left side, respectively

Figure 3 The proposed route for the deacetylation reaction of 3 under methanolic ammonia.
Table 1. Yields of the major products and reaction conditions of the de-O-acetylation reactions of compound 3 with NH3 (L) in CH3OH/THF (1:1) solution.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
