Table 1.
Optimizations of the reaction conditions.a
Citation:
Song Yanying, Wang Lili, Duan Zheng, Mathey François. Divergent intramolecular reactions between phosphines and alkynes[J]. Chinese Chemical Letters,
2020, 31(2): 329-332.
doi:
10.1016/j.cclet.2019.05.053

Divergent intramolecular reactions between phosphines and alkynes
English
Divergent intramolecular reactions between phosphines and alkynes
Abstract:
A divergent intramolecular reaction of phosphine tethered alkyne in protic solvent was developed. This provided a novel and simple access to a large variety of (Z)-alkenylphosphine oxides and phospholane oxides. Our preliminary studies suggested that these divergent reactions are closely related to the reaction condition and molecular structure. A possible mechanism of C-P bond cleavage of a pentacoordinated hydroxyphosphorane intermediate was proposed.
-
Key words:
- C-P bond cleavage
- / Phosphine
- / Alkyne
- / Alkenylphosphine
- / Phospholane
-
The carbon-carbon triple bond appears to be extremely versatile due to its ability to undergo a wide variety of transformations for applications in medical chemistry, agrochemistry, and material chemistry [1]. There are numerous publications dealing with the synthetic applications of the C≡C bond [2].
Very recently, much attention has been paid to the synthesis of various important organophosphorus derivatives through phosphination or phosphorylation of carbon-carbon triple bond [3-5]. Among these useful transformations, the generation of active phosphorus species via selective C-P cleavage is a challenging topic. Transition-metal-catalyzed C-P bond cleavage plays an important role in the synthetic applications of diverse organophosphorus moieties [6]. Herein, we would like to report a rare example of the synthesis of Z-alkenylphosphine oxides and phospholane oxides via C-P bond cleavage in protic solvent.
During our explorations of the reaction with phosphine tethered alkyne [4], we tried to synthesize a terminal alkyne 2 from 1a by removing the trimethylsilyl (TMS) group. Interestingly, after 1a was stirred with K2CO3 (1 equiv.) at room temperature in a mixture of MeOH/DCM, an (Z)-alkenylphosphine oxide 3a was isolated, while the desired terminal alkyne 2 was not detected (Table 1, entry 1). A similar result was observed with Cs2CO3 (entry 2). Absence of base resulted in no conversion of 1a (entry 3).Increasing the amount of K2CO3 (entry 4) or the reaction temperature (entry 5) did not improve the yield of the reaction.We were pleased to observe the transformation of 1a even with a catalytic amount of K2CO3 (5 mol%, entry 7). When the mixture of H2O/DMSO was used as the solvent, the conversion of 1a proceeded very slowly in the presence of a catalytic amount of K2CO3. Surprisingly, phospholane oxide 4a was obtained as major product and only trace amount of 3a was observed (entry 8). Such phospholane could potentially be used as ligand [7] and organocatalyst [8] in organic synthesis, which is difficult to obtain by other methods [9]. The reaction efficiency could be increased at elevated temperature (entry 9). A higher amount of base did not improve the reaction efficiency (entry 10). The yield of 4a could be further improved by using KOTMS as the catalyst. More details can be found in Supporting information.
Table 1
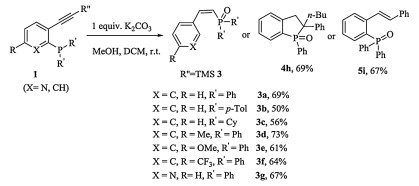
Encouraged by these unexpected results, we decided to investigate the synthetic applications of these base-promoted transformations of phosphine tethered alkynes in protic solvent.Our initial studies focused on the K2CO3 promoted transformation of phosphine tethered alkynes 1 in the mixture of MeOH/DCM. The reaction proved to be robust and versatile and allowed the synthesis of a broad range of (Z)-alkenylphosphine oxides 3 (Scheme 1). Both aryl-(3a, b) and alkyl-(3c) substituted phosphine oxides were well tolerated. Next, products with various substituted anchors were synthesized in modest to good yields, indicating that the reactions are essentially independent of the electronic properties of the anchors (3d, e: electron-donating Me and OMe; 3f: electron-withdrawing CF3). When the reaction was performed with a pyridine derivative 1g, 3g was isolated in 67% yield. To demonstrate the synthetic value of our newly developed reaction, the TMS group was replaced by a phenyl group. However, an alkyne reduced product 5i was obtained, which is in accordance with the reported results [10]. When n-butyl substituted 1h was used, phospholane oxide 4h was isolated as major product and a trace amount of unknown mixture of organophosphorus compounds was also observed. These results indicated the presence of TMS group is crucial to the formation of (Z)-alkenylphosphine oxides.
Scheme 1
 Scheme 1. The reaction of phosphine tethered alkyne in the presence of K2CO3. Reaction conditions: 1 (2 mmol), K2CO3 (2 mmol) in DCM (4 mL) and MeOH (4 mL), N2, room temperature, isolated yield.
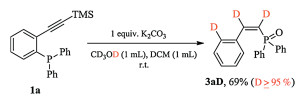
Scheme 1. The reaction of phosphine tethered alkyne in the presence of K2CO3. Reaction conditions: 1 (2 mmol), K2CO3 (2 mmol) in DCM (4 mL) and MeOH (4 mL), N2, room temperature, isolated yield.To be noticed, when the control reaction of 1a was performed in the deuterated solvent, methanol-d4/DCM, poly-deuterated product 3aD (D≥95%) was furnished (Scheme 2).
Scheme 2
 Scheme 2. Deuterated experiment.
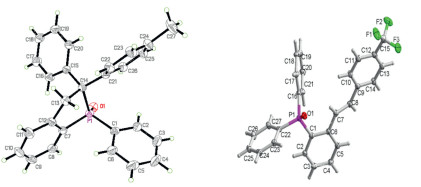
Scheme 2. Deuterated experiment.After screening the scope of (Z)-alkenylphosphine oxide, we moved our attention to the KOTMS catalyzed reaction of phosphine tethered alkynes 1 (Table 2). It is noteworthy that the complexity may arise with the TMS substituted alkynes (R'' = TMS), since the selectivity of the reactions is highly reliable on the substituents on phosphorus and the electronic effect of the aryl anchor. The diphenylphosphine derivative 1a gave the cyclized product 4a as major product (entry 1), while (Z)-alkenylphosphine oxide 3c was obtained selectively in the case of the dialkyl substituted phosphine 1c (entry 2). When R'' is TMS, the product will be either 3 or 4 (entries 1–5). The effect of R on anchored aryl group is elusive. When R is neutral H or Me, the formation of cyclized product 4 was favored (entries 1 and 3). When R is electrondonating OMe or electron-withdrawing CF3, (Z)-alkenylphosphine oxides were isolated as major products (entries 4 and 5). When R'' is aryl or alkyl group, the reactions gave 4 and/or 5 (entries 6–11). When R'' is electron neutral or rich aryl group, the formation of cyclized product 4 was preferred (entries 6 and 7). The structure of 4k was unambiguously established by X-ray crystallographic analysis (Fig. 1) (CCDC 1892220). The formation of 5 was preferred with electron deficient derivatives (entries 8 and 9). The structure of 5 m (CCDC 1892217) was confirmed by X-ray analysis (Fig. 1). The reaction of n-butyl substituted (R'' = n-Bu) 1 h gave cyclized 4 h, but the reaction of bulky alkyl substituted (R'' = t-Bu) 1n provided 5n.
Table 2
Figure 1
 Figure 1. The crystal structures of 4k and 5m.
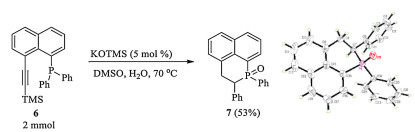
Figure 1. The crystal structures of 4k and 5m.The KOTMS catalyzed cascaded phosphination, aryl migration and protonation were used to synthesize a new fused phosphahexacycle 7 (Scheme 3).
Scheme 3
 Scheme 3. Synthesis of phosphahexacycle 7 (CCDC: 1892219).
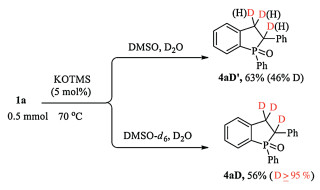
Scheme 3. Synthesis of phosphahexacycle 7 (CCDC: 1892219).To understand the reaction mechanism, the control and deuterium-labeling experiments were performed (Scheme 4).Partial deuterated product 4aD' was obtained in D2O/DMSO. The deuterated product 4aD (D≥95%) was isolated when the reaction was performed in D2O/DMSO-d6. These results indicated the importance of both H2O and DMSO in this tandem process.
Scheme 4
 Scheme 4. Control and deuterated experiments.
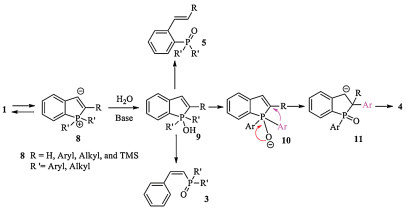
Scheme 4. Control and deuterated experiments.On the basis of the literature [11] and control experiments, a plausible reaction mechanism is proposed and outlined in Fig. 2. The reaction starts with an intramolecular nucleophilic attack of the alkynyl group by phosphine to form zwitterionic 8 [10]. Subsequently, hydrolysis of 8 by the protic solvent to generate pentacoordinated hydroxyphosphorane 9, followed by the decomposition of 9 to give the tertiary phosphine oxide 3 or 5. Upon the removal of the hydroxyl proton from 9, a concerted aryl migration process gives 11 [12], which provides phospholane oxide 4 after hydrolysis.
Figure 2
 Figure 2. The proposed mechanism.
Figure 2. The proposed mechanism.In summary, a divergent reaction of phosphine tethered alkyne in protic solvent has been realized. Our preliminary studies suggest that these divergent reactions are closely related to the reaction condition and molecular structure. In the presence of K2CO3, the TMS substituted alkynes were converted into (Z)-alkenylphosphine oxides in the mixture of MeOH and DCM. The reaction pathways in the mixture of H2O and DMSO with a catalytic amount of KOTMS are complicated. The divergences are dependent on the electronic nature of aryl anchor, the substituents on alkyne and phosphine: When TMS substituted alkynes were used, the alkylphosphines gave (Z)-alkenylphosphine oxides selectively. Arylphophines with electron neutral anchors produced phospholane oxides as major products, which are difficult to access by other methods. Arylphophines with electron-rich or deficient anchor preferred the formation of (Z)-alkenylphosphine oxides. When aryl substituted alkynes were used, the electron-rich aryl substituents favored the formation of phospholane oxides while the electrondeficient ones favored the alkenylphosphine oxide products. Placement of primary alkyl group on the end of alkyne allows for the selective formation of phospholane oxide. But the bulky alkyl substituted alkyne gives the alkenylphosphine oxide. Further efforts on the understanding the rules governing the selective transformations of phosphine tethered alkynes as well as the synthetic applications of these transformations are underway in our laboratory.
Acknowledgments
We thank the National Natural Science Foundation of China (Nos. 21672193, 21272218) and Henan Province postdoctoral research start-up project and Zhengzhou University of China for financial support of this research.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.053.
-
-
[1]
(a) B.M. Trost, C.J. Li, Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, Wiley-VCH, Weinheim, 2014, pp. 1-6;
(b) P.J. Stang, F. Diederich, Modern Acetylene Chemistry, Wiley, New York, 2008,
(c) F. Diederich, P.J. Stang, R.R. Tykwinski, Acetylene chemistry, Chemistry, Biology and Material Science, Wiley-VCH, Weinheim, 2005. -
[2]
(a) N. Sauermann, T.H. Meyer, Y. Qiu, et al., ACS Catal. 8 (2018) 7086-7103;
(b) S.J. Hein, D. Lehnherr, H. Arslan, et al., Acc. Chem. Res. 50 (2017) 2776-2788;
(c) I.V. Alabugin, E. Gonzalez-Rodriguez, Acc. Chem. Res. 51 (2018) 1206-1219;
(d) T. Michinobu, F. Diederich, Angew. Chem. Int. Ed. 57 (2018) 3552-3577;
(e) Y.Y. Hu, C.Y. Wang, ChemCatChem 11 (2019) 1167-1174;
(f) M. Patel, R.K. Saunthwal, A.K. Verma, Acc. Chem. Res. 50 (2017) 240-254;
(g) K. Lauder, A. Toscani, N. Scalacci, et al., Chem. Rev.117 (2017) 14091-14200;
(h) B. Prabagar, N. Ghosh, A.K. Sahoo, Synlett. 28 (2017) 2539-2555;
(i) B.M. Trost, J.T. Masters, Chem. Soc. Rev. 45 (2016) 2212-2238. -
[3]
(a) T.Q. Chen, C.Q. Zhao, L.B. Han, J. Am. Chem. Soc. 140 (2018) 3139-3155;
(b) T.Z. Huang, Y. Saga, H.Q. Guo, et al., J. Org. Chem. 83 (2018) 8743-8749;
(c) H.M. Wang, Y.L. Li, Z.L. Tang, et al., ACS Catal. 8 (2018) 10599-10605;
(d) L.L. Khemchyan, J.V. Ivanova, S.S. Zalesskiy, et al., Adv. Synth. Catal. 356 (2014) 771-780;
(e) R.W. Shen, J.L. Yang, M. Zhang, et al., Adv. Synth. Catal. 359 (2017) 3626-3637;
(f) S.M.Härling, B.E.Fener, S.Krieck, etal., Organometallics37 (2018)4380-4386;
(g) P.Y. Geant, J.P. Uttaro, S. Peyrottes, et al., Eur. J. Org. Chem. (2017) 3850-3855;
(h)H.Ohmiya, H.Yorimitsu, K.Oshima, Angew.Chem.Int.Ed.44 (2005)2368-2370;
(i) P.H. Lee, S. Kim, A. Park, et al., Angew. Chem. Int. Ed. 49 (2010) 6806-6809;
(j) M.Y. Niu, H. Fu, Y.Y. Jiang, et al., Chem. Commun. (2007) 272-274;
(k)P.Nun, J.D.Egbert, M.J.Oliva-Madrid, etal., Chem.-Eur.J.18 (2012)1064-1067;
(l) M. Hayashi, Y. Matsuura, Y. Watanabe, J. Org. Chem. 71 (2006) 9248-9251;
(m) L.B. Han, M. Tanaka, J. Am. Chem. Soc. 118 (1996) 1571-1572;
(n) L.B. Han, C. Zhang, H. Yazawa, et al., J. Am. Chem. Soc.126 (2004) 5080-5081;
(o)C.Q.Zhao, L.B.Han, M.Goto, etal., Angew.Chem.Int.Ed.40 (2001)1929-1932;
(p) I.G. Trostyanskaya, I.P. Beletskaya, Tetrahedron 70 (2014) 2556-2562;
(q) V.P. Ananikov, J.V. Ivanova, L.L. Khemchyan, et al., Eur. J. Org. Chem. (2012) 3830-3840. -
[4]
(a) Y.Z. Xu, Z.H. Wang, Z.J. Gan, et al., Org. Lett. 17 (2015) 1732-1734;
(b) Y. Zhou, Z.J. Gan, B. Su, et al., Org. Lett. 17 (2015) 5722-5724;
(c) X. Zhao, Z.M. Lu, Q.Y. Wang, et al., Organometallics 35 (2016) 3440-3443;
(d) X. Zhao, Z.J. Gan, C.P. Hu, et al., Org. Lett. 19 (2017) 5814-5817;
(e) J.J. Hou, Y.Z. Xu, Z.J. Gan, et al., J. Organomet. Chem. 879 (2019) 158-161;
(f) G.Y. Tao, Z. Duan, F. Mathey, Org. Lett. 21 (2019) 2273-2276. -
[5]
(a) A.Y. Peng, Y.X. Ding, J. Am. Chem. Soc. 125 (2003) 15006-15007;
(b) H. Tsuji, K. Sato, L. Ilies, et al., Org. Lett. 10 (2008) 2263-2265;
(c) T. Sanji, K. Shiraishi, T. Kashiwabara, et al., Org. Lett. 10 (2008) 2689-2692;
(d) Y.R. Chen, W.L. Duan, J. Am. Chem. Soc. 135 (2013) 16754-16757;
(e) B. Pérez-Saavedra, N. Vázquez-Galiñanes, C. Saá, et al., ACS Catal. 7 (2017) 6104-6109;
(f) A.Fukazawa, E.Yamaguchi, E.Ito, etal., Organometallics30 (2011)3870-3879;
(g)M.Arribat, E.Rémond, S.Clément, etal., J.Am.Chem.Soc.140 (2018)1028-1034;
(h)S.Arndt, M.M.Hansmann, F.Rominger, etal., Chem.-Eur.J.23 (2017)5429-5433;
(i) C.G. Wang, A. Fukazawa, Y. Tanabe, et al., Chem. -Asian J.13 (2018) 1616-1624. -
[6]
(a) P.E. Garrou, Chem. Rev. 85 (1985) 171-185;
(b) A.W. Parkins, Coord. Chem. Rev. 250 (2006) 449-467;
(c) S.A. Macgregor, Chem. Soc. Rev. 36 (2007) 67-76;
(d) L.L. Wang, H. Chen, Z. Duan, Chem. -Asian J. 13 (2018) 2164-2173;
(e) Y.H. Lee, B. Morandi, Coord. Chem. Rev. 386 (2019) 96-118. -
[7]
(a) X.W. Zhang, K.X. Huang, G.H. Hou, et al., Angew. Chem. Int. Ed. 49 (2010) 6421-6424;
(b) D. Liu, X.M. Zhang, Eur. J. Org. Chem. (2005) 646-649;
(c) W.J. Tang, X.M. Zhang, Angew. Chem. Int. Ed. 41 (2002) 1612-1614;
(d) H. Shimizu, T. Saito, H. Kumobayashi, Adv. Synth. Catal. 345 (2003) 185-189;
(e) B. Song, C.B. Yu, W.X. Huang, et al., Org. Lett. 17 (2015) 190-193;
(f) H.L. Yang, E.F. Wang, P. Yang, et al., Org. Lett.19 (2017) 5062-5065;
(g) Z.W. Lu, H.Y. Zhang, Z.P. Yang, et al., ACS Catal. 9 (2019) 1457-1463. -
[8]
(a) C.J. O'Brien, J.L. Tellez, Z.S. Nixon, et al., Angew. Chem. Int. Ed. 48 (2009) 6836-6839;
(b) D.J.Carr, J.S.Kudavalli, K.S.Dunne, etal., J.Org.Chem.78 (2013)10500-10505;
(c)Y.M.Xiao, Z.H.Sun, H.C.Guo, etal., BeilsteinJ.Org.Chem.10 (2014)2089-2121. -
[9]
(a) A.M.Gregson, S.M.Wales, S.J.Bailey, etal., J.Org.Chem.80 (2015)9774-9780;
(b) D.J. Collins, L.E. Rowley, J.M. Swan, Aust. J. Chem. 27 (1974) 831-839;
(c) Y.Q. Zhou, X.Y. Yan, C.J. Xi, Tetrahedron Lett. 51 (2010) 6136-6138;
(d) K. Wlodarczyk, M. Stankevic, Tetrahedron 72 (2016) 5074-5090;
(e) F.G. Mann, I. Milla, J. Chem. Soc. (1951) 2205-2206;
(f)W.L.Orton, K.A.Mesch, L.D.Quin, PhosphorusSulfurRelat.Elem.5 (1979)349-357;
(g) C.H. Chen, K.E. Brighty, Tetrahedron Lett. 21 (1980) 4421-4424;
(h)S.Shah, M.C.Simpson, R.C.Smith, etal., J.Am.Chem.Soc.123 (2001)6925-6926;
(i)H.H.Karsch, H.U.Reisacher, PhosphorusSulfurRelat.Elem.36 (1988)213-215;
(j) T. Imamoto, T. Yoshizawa, K. Hirose, et al., Heter. Chem. 6 (1995) 99-104;
(k) T.J. Brunker, B.J. Anderson, N.F. Blank, et al., Org. Lett. 9 (2007) 1109-1112. -
[10]
(a) W. Winter, Angew. Chem. Int. Ed. 17 (1978) 947-948;
(b) T. Butters, I. Haller-Pauls, W. Winter, Chem. Ber. 115 (1982) 578-592. -
[11]
(a) G.W. Penton, C.K. Ingold, J. Chem. Soc. (1929) 2342-2357;
(b) L. Horner, H. Hoffmann, H.G. Wippel, et al., Chem. Ber. 91 (1958) 52-57;
(c) M. Zanger, C.A. Wander Werf, W.E. McEwen, J. Am. Chem. Soc. 81 (1959) 3806-3807;
(d) G. Aksnes, J. Songstad, Acta Chem. Scand. 16 (1962) 1426-1432;
(e) R.F. Hudson, M. Green, Angew. Chem. Int. Ed. 2 (1963) 11-20;
(f) W.E. McEwen, K.F. Kumli, A. Blade-Font, et al., J. Am. Chem. Soc. 86 (1964) 2378-2384;
(g) J.R. Corfield, S. Trippett, Chem. Commun. 19 (1970) 1267;
(h) D.A. Allen, S.J. Grayson, I. Harness, et al., J. Chem. Soc. Perkin Trans. I 2(14) (1973) 1912-1915;
(i) A. Schnell, J.C. Tebby, J. Chem. Soc. Perkin Trans. I 1 (1977) 1883-1886;
(j)R.A.McClelland, G.H.McGall, G.Patel, J.Am.Chem.Soc.107 (1985)5204-5209;
(k) D.G. Gilheany, N.T. Thompson, B.J. Walker, Tetrahedron Lett. 28 (1987) 3843-3844;
(l)H.J.Cristau, P.Mouchet, PhosphorusSulfurSiliconRelat.Elem.107 (1995)135-144;
(m) K.Y. Lee, J.E. Na, M.J. Lee, et al., Tetrahedron Lett. 45 (2004) 5977-5981. -
[12]
B. Li, M.K. Zhang, X.L. Huang, et al., Org. Chem. Front. 4 (2017) 1854-1857. doi: 10.1039/C7QO00310B
-
[1]
-
Scheme 1 The reaction of phosphine tethered alkyne in the presence of K2CO3. Reaction conditions: 1 (2 mmol), K2CO3 (2 mmol) in DCM (4 mL) and MeOH (4 mL), N2, room temperature, isolated yield.
-

 DownLoad:
DownLoad:



 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 9
- 文章访问数: 2297
- HTML全文浏览量: 178
