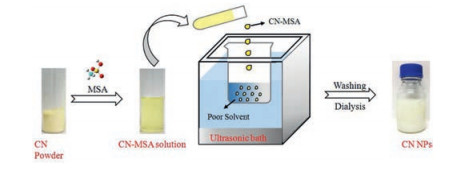
Figure 1.
Illustration of CN NPs preparation.
Preparation of carbon nitride nanoparticles by nanoprecipitation method with high yield and enhanced photocatalytic activity
Ziyu Gan , Chaofeng Huang , Yanfei Shen , Qing Zhou , Dan Han , Jin Ma , Songqin Liu , Yuanjian Zhang

As a promising 2D polymer material, graphite carbon nitride (CN) has been widely studied in the fields of solar energy conversion [1-3], environmental remediation [4-7] and optoelectronic biosensing [8-10]. This is ascribed to its appealing properties, such as tunable electronic band structure, excellent physicochemical stability and facile synthesis from inexpensive and abundant raw materials [11-15]. Engineering of micro/nano structure of bulk CN has been generally utilized to improve the performance. For instance, several pioneering works regarding convert bulk CN into different nanostructures have been reported by template methods [16-20], supramolecular preorganization approach [21-23], liquid exfoliation [8, 24, 25] and other chemical tailoring methods [26-28]. These strategies well controlled the structural properties and interlayer interactions of CN, thus effectively improving specific surface area, prolonging lifetime of charge carriers, enhancing electron transport and separation efficiency. As a result, the photocatalytic and optoelectronic performances of CN in end-applications were greatly boosted.
Nevertheless, there are still some limitations that need to be addressed before the large-scale industrial applications of CN [29, 30]. For example, the hard template method for preparing mesoporous CN and CN nanoparticles (NPs) suffers from the use of hazardous etchants including NH4HF2/HF, and is often timeconsuming. The soft template method often leads to the residual carbon impurities that may act as carrier recombination centers and lower catalytic activity. The liquid exfoliation strategy usually requires a long time of sonication treatment (typically 10–16 h) in organic solvent or water, and the as-obtained suspensions of CN nanosheets has a relatively low concentration (0.15 mg/mL) [8, 24]. Therefore, seeking a simple and efficient method for nanostructured CN with high yield would be fascinating for the subsequent research of CN in large-scale industrial applications.
Here, we report the preparation of CN NPs by a nanoprecipitation approach [31]. The CN NPs could be facilely prepared by simple dissolution of CN in methanesulfonic acid (MSA) and precipitation using poor solvents (Fig. 1), with a yield as high as 50%, the highest one to the best of our knowledge. Moreover, the as-prepared CN NPs were successfully applied to photocatalytic degradation of dyes with an evident boosted performance up to 2.5 times with respect to the pristine bulk CN.
Nanoprecipitation technique has been widely used for the polymer nanoparticles preparation in the arena of pharmaceutical, agricultural, food and cosmetic [32-34, 31]. This technique is mainly based on dissolution and precipitation mechanism. It has the advantages of simplicity, low cost and energy input, easy to scale-up, satisfying reproducibility and obtaining of micro/nano particle sizes with narrow size distribution. The pre-requirements for the successful nanoprecipitation largely rely on the distinct solubility of the solute (i.e., the bulk precursor) between good and poor solvents. However, bulk CN is almost insoluble in most solvents, which greatly hinder this intriguing approach for engineering the nanostructure of CN. Our group recently discovered that the sulfonic group containing solvents such as concentrated sulfuric acid and methanesulfonic acid (MSA) are two good solvents for dissolution of CN via sophisticated protonation and intercalation [35, 36]. Compared to concentrated sulfuric acid, MSA is much less oxidative, thus commonly used as a green solvent for many organic reactions. Therefore, it is highly feasible that MSA may be used as a good solvent to drive the nanoprecipitation process in preparation of nanostructure CN.
In general, some factors should be considered in choosing poor solvent for the proposed nanoprecipitation. An ideal poor solvent is the one, which is miscible with MSA but cannot dissolve CN and does not initiate any side reactions among itself, MSA and CN. In these regards, in the first set of experiments, bulk CN was dissolved in MSA to form a CN-MSA solution, and then precipitated by using acetonitrile as poor solvent. The precipitation process mainly consists of the following steps: generation of supersaturation, nucleation, growth by condensation, and coagulation [31, 32]. Supersaturation occurs when the solubility of CN decreased with the addition of poor solvent such that the concentration of dissolved CN in the solution exceeded the equilibrium saturation value. Subsequently, nucleation was induced when the supersaturation concentration of the system reached a certain critical level. When CN concentration decreased below the critical supersaturation concentration, nucleation stopped while the formed nuclei continued to grow bycondensation and coagulation. Condensation and coagulation are two different driving forces for CN growth. The mechanism of condensation is the addition of single molecules to the particle surface while the coagulation is mainly the collision of particles and then adhere to each other. In these regards, the ultrasonic treatment was often used as a disturbance to accelerate nano-precipitation process, inhibit the coagulation and prevent the formation of large CN aggregates [33, 34]. Moreover, it was observed that the raw CN NPs tended to agglomerate due to the high surface energy, as once the ultrasound was stopped, the resulting dispersed CN quickly flocculated. Nevertheless, after a through washing with water, the stable dispersion of CN would form again, which demonstratedthat thewelldispersionof CN was strongly depended on the acid strength and ion concentration [37]. The zeta potential of the CN dispersed was about 38.6 mV, which might be ascribing to the protonation effect by MSA [38, 35, 36]. Benefiting from the highly positive charged surface, the CN dispersion could keep stable without evident aggregation after standing for several weeks. Moreover, the aqueous solutions of CN NPs can also be stable in different pH ranges (Fig. S1 in Supporting information). In addition, the concentration of the CN dispersion by this method could reach up to 3 mg/mL, which was one order of magnitude higher than the previously reported concentration of stable CN dispersion in water or organic solvents (typically 0.15 mg/mL) [8].
To characterize the morphology and size of the as-prepared CN nanostructures, the measurements of scanning electron microscopy (SEM), transmission electron microscopy (TEM), and dynamic light scattering (DLS) measurements were conducted. The SEM image in Fig. 2a showed bulk CN randomly consisted of large size particles of several micrometers with lamellar texture. In contrast, after nanoprecipitation, it turned into uniform nanoparticles and dramatically decreased to several ten nanometers (Fig. 2b). The TEM image of CN NPs in higher magnification further demonstrated that these CN NPs was thin, reminiscent of stacked layer structure of pristine bulk CN (Fig. 2c). The average size of CN NPs was estimated to be ca. 36 nm by using SEM images of CN particles (Fig. 2d, right panel). In addition, DLS measurement showed that the lateral size of CN NPs was around 37 nm (Fig. 2d, left panel), consisted with the statistical results of SEM observation. Notably, the morphology of CN NPs obtained in this study was not a perfect circular shape, which might be attributed to the ultrasonic assisted synthesis approach used for the CN NPs preparation.

To gain insight into the texture properties of CN NPs, the crystal structure of bulk CN and CN NPs were studied by X-ray diffraction (XRD) method. In general, the XRD pattern in Fig. 3a show that the CN NPs gave similar two consistent diffraction peaks with bulk CN: one was the peak (002) at about 27.4°, originating from the graphitic interlayer structure and the other was the peak (100) at about 13.1°, deriving from in-planar repeated structure units [8, 39, 40]. It verified that the basic lamellar CN crystal structure was largely retained. Noticeably, nevertheless, in contrast to bulk CN, the intensity of these two peaks for CN NPs both sharply decreased, mainly ascribing to the decreased planar size of the CN layers and the reduced periodic interlayer stacking. Moreover, it was noted that the full-width at half maximum (FWHM) of CN nanoparticles (1.55°) from the most prominent peak (002) increased with respected to that of bulk CN (1.26°), another evidence of the deceased crystalline size. These phenomena well supported the fact of successful preparation of CN NPs after nanoprecipitation processes, which was also consistent well with the results in TEM and SEM observation.
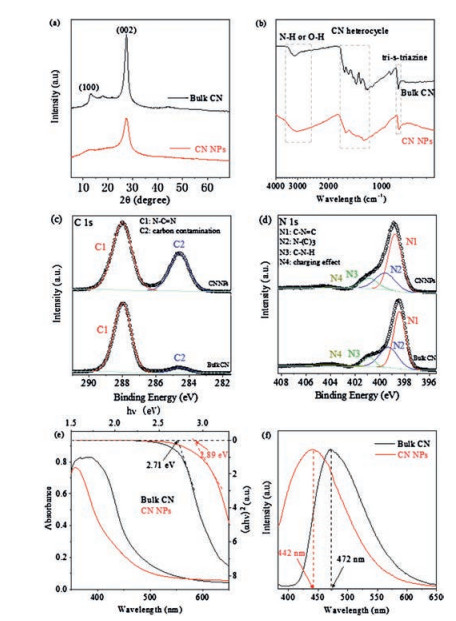
The chemical structure of CN NPs was characterized by using Fourier transform infrared (FT-IR) spectra. Fig. 3b showed that the spectrum of CN NPs was similar to that of bulk CN, both of which had the sharp breathing mode peak of the tri-s-triazine units at ~801 cm-1. Besides, the typical stretching modes bands of aromatic CN heterocycles at 1200–1650 cm-1 and the N—H stretching vibration peak at 3000–3500 cm-1 were observed [41]. Therefore, the chemical structure of bulk CN was mostly retained after the transformation to CN NPs. At the same time, some minor changes in the CN NPs spectrum was also observed. For example, the N—H stretching vibration peak became much broader, and some of the characteristic peaks shifted slightly to the low wavelength region. Moreover, the relative intensity change of CN heterocycles absorption peaks was also noticed. These phenomena were observed in previous studies and could be explained by the protonation by MSA [38, 35, 36]. To gain insight into the more detailed bonding characteristics of CN NPs, the X-ray photoelectron spectra (XPS) were measured. The C 1 s XPS spectra of CN NPs and bulk CN (Fig. 3c) both showed two similar main peaks. The predominant C1 peak at 288 eV originated from N—C = N in the typical aromatic ring [39, 42, 43], suggesting that the CN NPs had the same backbone as the pristine bulk sample. Meanwhile, the spectrum of N 1s for bulk CN (Fig. 3d) could be deconvoluted into four peaks at 398.4, 399.3, 400.7 and 404.0 eV, which were attributed to C=N—C (tri-s-triazine rings), N—(C)3 (bridging N atoms), C—N—H (bonded with H atoms) and the charge effect, respectively [36, 44]. This phenomenon was also observed for CN NPs, further verifying the similar C—N bonding characteristics of these two compounds. Besides, the O 1s and S 2p XPS spectra (Figs. S2a and b in Supporting information) of CN NPs demonstrated changes, compared to that of bulk CN, which could be due to the traces of methanesulfonic acid on the surface via protonation interaction [35, 36, 38].
The electronic structure and optical properties of as-prepared CN NPs were further studied by UV–vis diffuse reflectance and photoluminescence (PL) spectra. In contrast to bulk CN, the UV–vis absorption spectrum (Fig. 3e) of CN NPs showed a slight blue shift and the band gap increased from ~2.71 eV to 2.89 eV. Additionally, the larger bandgap of the CN NPs was confirmed by the blue shift of the PL spectrum (Fig. 3f and Fig. S3 in Supporting information). The slight blue shifts in the spectra of CN NPs in comparison with bulk CN should be mainly ascribed to the quantum confinement effect of decreased size of CN NPs and the protonation by MSA, leading to the valence band and conduction band position move in opposite directions [9, 35, 36, 38].
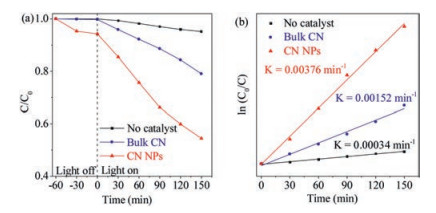
The photocatalytic performance of CN NPs was investigated by degradation of MB under full light irradiation (Fig. 4a and Fig. S4 in Supporting information). After irradiation for 150 min, MB was photocatalytically degradated more than 40%, much faster than by pristine bulk CN. In contrast, MB molecules were stable and almost no significant degradation occurred under the identical illumination conditions, indicating that the catalytic activity was derived from the photocatalysts. Moreover, the first-order constant (k) for photocatalytic MB degradation of bulk CN and CN NPs was calculated by ln(C0/Ct) = kt. The time-course variation of ln(C0/Ct) is shown in Fig. 4b. The kinetic constants of bulk CN and CN NPs were 0.0015 min-1 and 0.0037 min-1, respectively. The result showed that the as-prepared CN NPs demonstrated an boosted photocatalytic activity up to 2.5 times in degradation of MB dye.
It should be noted that the proposed nano-precipitation strategy was also applicable by using other poor solvents for bulk CN. For instance, methanol was also selected and investigated in preparation of CN nanoparticles, which demonstrated similar results (Figs. S5-S11 in Supporting information). Nevertheless, more studies are still needed to explore the detailed nanoprecipitation kinetics for CN NPs using different solvents and other control conditions as well to further modulate the morphology and surface properties of the as-obtained CN nanoparticles.
In summary, we reported a simple and fast method for CN NPs preparation via a nanoprecipitation process based on the recent discovery of good solvents for bulk CN. This method, which mainly consisted of CN dissolution and precipitation, could efficiently obtain highly dispersed CN NPs (~40 nm) with a yield of up to 50%, the highest one compared to previous top-down methods for nanostructured CN preparation to our knowledge, such as liquidstate exfoliation. Moreover, the as-prepared CN NPs showed the improved photocatalytic degradation of dye activity due to its smaller sizes. These results revealed the potential of cost-effective batch preparation of nanostructured CN and would expand the large-scale industrial applications of CN. Besides, the power of ultrasonic bath would be an important factor to further modulate the size of CN NPs and if the size of CN NPs was effective engineered, it is highly expected to further measure the intriguing quantum confine effect [45, 46]. Work focused this speculation is ongoing.
This work was financially supported in part by the National Natural Science Foundation of China (Nos. 21775018, 21675022), the Natural Science Foundation of Jiangsu Province (Nos. BK20160028, BK20170084), the Open Funds of the State Key Laboratoryof Electroanalytical Chemistry (No.SKLEAC201909) and the Fundamental Research Funds for the Central Universities.
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.065.
X. Wang, K. Maeda, A. Thomas, et al., Nat. Mater. 8(2009) 76-80. doi: 10.1038/nmat2317
Y. Zhang, T. Mori, J. Ye, M. Antonietti, J. Am. Chem. Soc.132(2010) 6294-6295. doi: 10.1021/ja101749y
J. Liu, Y. Liu, N. Liu, et al., Science 347(2015) 970-974. doi: 10.1126/science.aaa3145
S. Yan, Z. Li, Z. Zou, Langmuir 25(2009) 10397-10401. doi: 10.1021/la900923z
W.J. Ong, J.J. Yeong, L.L. Tan, et al., RSC. Adv. 4(2014) 59676-59685. doi: 10.1039/C4RA10467F
P.X. Qiu, H. Chen, C.M. Xu, et al., J. Mater. Chem. A 3(2015) 24237-24244. doi: 10.1039/C5TA08406G
J. Wang, Y. Shen, Y. Li, S. Liu, Y. Zhang, Chem. -Eur. J. 22(2016) 12449-12454. doi: 10.1002/chem.201602095
X. Zhang, X. Xie, H. Wang, et al., J. Am. Chem. Soc. 135(2012) 18-21.
J. Ji, J. Wen, Y. Shen, et al., J. Am. Chem. Soc. 139(2017) 11698-11701. doi: 10.1021/jacs.7b06708
Y. Lv, S. Chen, Y. Shen, et al., J. Am. Chem. Soc. 140(2018) 2801-2804. doi: 10.1021/jacs.8b00515
G. Liu, P. Niu, C. Sun, et al., J. Am. Chem. Soc. 132(2010) 11642-11648. doi: 10.1021/ja103798k
B. Cai, X. Lv, S. Gan, et al., Nanoscale 5(2013) 1910-1916. doi: 10.1039/c2nr33521b
H. Li, S. Gan, H. Wang, D. Han, L. Niu, Adv. Mater. 27(2015) 6906-6913. doi: 10.1002/adma.201502755
Y. Chen, Z. Zhan, J. Wang, et al., Chin. Chem. Lett. 29(2018) 437-440. doi: 10.1016/j.cclet.2017.08.028
M. Liu, P. Xia, L. Zhang, B. Cheng, J. Yu, ACS Sustain. Chem. Eng. 6(2018) 10472-10480. doi: 10.1021/acssuschemeng.8b01835
M. Groenewolt, M. Antonietti, Adv. Mater. 17(2005) 1789-1792. doi: 10.1002/adma.200401756
F. Goettmann, A. Thomas, M. Antonietti, Angew. Chem. Int. Ed. 46(2007)2717-2720. doi: 10.1002/anie.200603478
A. Vinu, Adv. Funct. Mater. 18(2008) 816-827. doi: 10.1002/adfm.200700783
X.H. Li, J. Zhang, X. Chen, et al., Chem. Mater. 23(2011) 4344-4348. doi: 10.1021/cm201688v
J. Wang, C. Zhang, Y. Shen, et al., J. Mater. Chem. A 3(2015) 5126-5131. doi: 10.1039/C4TA06778A
Y.S. Jun, E.Z. Lee, X. Wang, et al., Adv. Funct. Mater. 23(2013) 3661-3667. doi: 10.1002/adfm.201203732
M. Shalom, S. Inal, C. Fettkenhauer, D. Neher, M. Antonietti, J. Am. Chem. Soc. 135(2013) 7118-7121. doi: 10.1021/ja402521s
Y. Ishida, L. Chabanne, M. Antonietti, M. Shalom, Langmuir 30(2014) 447-451. doi: 10.1021/la404101h
S. Yang, Y. Gong, J. Zhang, et al., Adv. Mater. 25(2013) 2452-2456. doi: 10.1002/adma.201204453
X. She, H. Xu, Y. Xu, et al., J. Mater. Chem. A 2(2014) 2563-2570. doi: 10.1039/c3ta13768f
J. Zhang, M. Zhang, L. Lin, X. Wang, Angew. Chem. 127(2015) 6395-6399. doi: 10.1002/ange.201501001
Z. Zhou, Y. Shen, Y. Li, et al., ACS Nano 9(2015) 12480-12487. doi: 10.1021/acsnano.5b05924
Y. Zhang, Z. Zhou, Y. Shen, et al., ACS Nano 10(2016) 9036-9043. doi: 10.1021/acsnano.6b05488
W.J. Ong, L.L. Tan, Y.H. Ng, S.T. Yong, S.P. Chai, Chem. Rev.116(2016) 7159-7329. doi: 10.1021/acs.chemrev.6b00075
Z.Zhou, Y. Zhang, Y.Shen, S. Liu, Y.Zhang, Chem.Soc.Rev.47(2018)2298-2321. doi: 10.1039/C7CS00840F
C.J.M. Rivas, M. Tarhini, W. Badri, et al., Int. J. Pharm. 532(2017) 66-81. doi: 10.1016/j.ijpharm.2017.08.064
I.J. Joye, D.J. McClements, Trends. Food. Sci. Tech. 34(2013) 109-123. doi: 10.1016/j.tifs.2013.10.002
J.F.Y. Fong, S.F. Chin, S.M. Ng, Sens. Actuators B: Chem. 209(2015) 997-1004. doi: 10.1016/j.snb.2014.12.038
Y. Chang, X. Yan, Q. Wang, et al., Carbohyd. Polym. 157(2017) 1413-1418. doi: 10.1016/j.carbpol.2016.11.019
Z. Zhou, J. Wang, J. Yu, et al., J. Am. Chem. Soc. 137(2015) 2179-2182. doi: 10.1021/ja512179x
C. Huang, J. Wen, Y. Shen, et al., Chem. Sci. 9(2018) 7912-7915. doi: 10.1039/C8SC03855D
F. Cheng, H. Wang, X. Dong, Chem. Commun. 51(2015) 7176-7179. doi: 10.1039/C5CC01035G
Y. Zhang, A. Thomas, M. Antonietti, X. Wang, J. Am. Chem. Soc.131(2008) 50-51.
P. Niu, L. Zhang, G. Liu, H.M. Cheng, Adv. Funct. Mater. 22(2012) 4763-4770. doi: 10.1002/adfm.201200922
C. Zhou, R. Shi, L. Shang, et al., Nano. Res. 11(2018) 3462-3468. doi: 10.1007/s12274-018-2003-2
Y.F. Shen, C. Zhang, C.G. Yan, H.Q. Chen, Y.J. Zhang, Chin. Chem. Lett. 28(2017) 1312-1317. doi: 10.1016/j.cclet.2017.04.004
V.W. Lau, M.B. Mesch, V. Duppel, et al., J. Am. Chem. Soc.137(2015) 1064-1072. doi: 10.1021/ja511802c
Q. Lin, L. Li, S. Liang, et al., Appl. Catal. B: Environ. 163(2015) 135-142. doi: 10.1016/j.apcatb.2014.07.053
Z. Zhou, J. Wang, J. Yu, et al., J. Am. Chem. Soc. 137(2015) 2179-2182. doi: 10.1021/ja512179x
T. Gholami, M. Salavati-Niasari, M. Bazarganipour, E. Noori, Superlattice. Microstruct. 61(2013) 33-41. doi: 10.1016/j.spmi.2013.06.004
V. Safarifard, A. Morsali, S.W. Joo, Ultrason. Sonochem. 20(2013) 1254-1260. doi: 10.1016/j.ultsonch.2013.01.014

Figure 2 SEM images of (a) pristine bulk CN and (b) CNNPs.(c)TEM image of CNNPs.(d) DLS size distribution of the CN NPs dispersion(leftpanel)and size distribution of the CN NPs (N = 100, right panel) calculated by using the SEM data in (b).

Figure 3 (a) XRD pattern, (b) FT-IR, high resolution (c) XPS C 1s and (d) N 1s spectra, (e) UV–vis, and (f) Normalized PL spectra of bulk CN and CN NPs powders.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
