Citation:
Zhou Yang, Fang Aiping, Wang Fazhan, Li Huili, Jin Quansheng, Huang Lingjing, Fu Chunmei, Zeng Jun, Jin Zhaohui, Song Xiangrong. Core-shell lipid-polymer nanoparticles as a promising ocular drug delivery system to treat glaucoma[J]. Chinese Chemical Letters,
2020, 31(2): 494-500.
doi:
10.1016/j.cclet.2019.04.048
Core-shell lipid-polymer nanoparticles as a promising ocular drug delivery system to treat glaucoma
English
Core-shell lipid-polymer nanoparticles as a promising ocular drug delivery system to treat glaucoma
State Key Laboratory of Biotherapy and Cancer Center/Department of Pharmacy, West China Hospital, and Collaborative Innovation Center for Biotherapy, Sichuan University, Chengdu 610041, China
b.
West China School of Pharmacy, Sichuan University, Chengdu 610041, China
songxr@scu.edu.cn(X. Song). 1 These authors contributed equally to this work.
Received Date:
28 February 2019 Accepted Date:
01 April 2019 Revised Date:
30 March 2019 Available Online:
01 February 2020
Abstract:
Nowadays, tremendous researches have been focused on the core-shell lipid-polymer nanoparticles (LPNs) due to the advantages of both liposomes and polymer nanoparticles. In this work, LPNs were applied to encapsulate brinzolamide (Brz-LPNs) for achieving sustained drug release, improving drug corneal permeation and enhancing drug topical therapeutic effect. The structure of Brz-LPNs was composed of poly(lactic-co-glycolic) acid (PLGA) nanocore which encapsulated Brz (Brz-NPs) and lipid shell around the core. Brz-LPNs were prepared by a modified thin-film dispersion method. With the parameters optimization of Brz-LPNs, optimal Brz-LPNs showed an average particle size of 151.23±1.64 nm with a high encapsulation efficiency (EE) of 86.7%±2.28%. The core-shell structure of Brz-LPNs were confirmed by transmission electronic microscopy (TEM). Fourier transformed infrared spectra (FTIR) analysis proved that Brz was successfully entrapped into Brz-LPNs. Brz-LPNs exhibited obvious sustained release of Brz, compared with AZOPT® and Brz-LPs. Furthermore, the corneal accumulative permeability of Brz-LPNs significantly increased compared to the commercial available formulation (AZOPT®) in vitro. Moreover, Brz-LPNs (1 mg/mL Brz) showed a more sustained and effective intraocular pressure (IOP) reduction than Brz-LPs (1 mg/mL) and AZOPT® (10 mg/mL Brz) in vivo. In conclusion, Brz-LPNs, as promising ocular drug delivery systems, are well worth developing in the future for glaucoma treatment.
With the fast development of nanotechnology, a large number of advantages of liposomes and nanoparticles have attracted numerous attention in the biomedical field [1]. As we all know, liposomes have been widely applied to drug delivery systems for decades [2, 3]. At present, some exploratory studies of liposomes for ocular delivery have been carried out owing to the superior drug absorption, biocompatible, biodegradable and nontoxic or mildly toxic [4]. From the literature, it is evident that liposomes can get to the retina when administered topically as eyedrops [5]. However, the widespread application of liposomes for ocular delivery is limited by some factors such as drug leakage, aggregation, instability during storage and short half-lives [6]. Therefore, more researches need to be conducted on the drug delivery systems to overcome the above shortcomings and obtain a more stable system. In addition, polymer nanoparticles are another promising platform for drug delivery [7]. It is well-known that PLGA was approved by Food and Drug Administration and European Medicines Agency because of its good biocompatibility, biodegradability, low toxicity [8]. Based on these outstanding properties, PLGA nanoparticles have been used in the domain of drug delivery with many advantages [9, 10]. According to published studies, PLGA nanoparticles have been used for ocular drug delivery [11-13], such as ocular anti-inflammatory applications via encapsulating carprofen or pranoprofen [14, 15], management of glaucoma by means of loading brinzolamide [16]. Therefore, the previous studies reveal that PLGA nanoparticles could be a promising ocular delivery system. However, lots of disadvantages of polymer nanoparticles remain to be resolved, such as drug leakage, polymer cytotoxicity, and polymer biocompatibility [17]. To address the aforementioned issues, a strong interest has been exerted on the exploration of novel ocular drug delivery system. With the further research on nanotechnology, core-shell type lipid-polymer nanoparticles (LPNs) have obtained tremendous attention on account of their special properties, which can mitigate some limitations associated with liposomes and polymeric nano-particles [18].
In recent years, considering the improvement in nanotechnol-ogy, LPNs have been developed after nanoparticles, liposomes and micelles for novel delivery vehicles [19]. It is very exciting that LPNs are composed of a polymer core and a phospholipid shell, resulting in LPNs possessing the superiorities of both polymer nanoparticles and liposomes [20]. The polymer core provides a stable skeletal structure with internal entrapped hydrophilic/hydrophobic drugs, and the phospholipid shell contribute to increased drug-embedding efficiency and good biocompatibility [21]. Briefly, LPNs, as a novel drug delivery system, have superior control release profile, high drug loading and suitable stability owing to the advantages of biodegradable polymeric nanoparticles and biomimetic liposomes. According to the literature, LPNs have been used for oral delivery of proteins and peptides [18, 22], systemic delivery for tumor-targeted therapy and etcetera [19, 23, 24]. In addition, some researches about core-shell for-mulations have been conducted on the ocular delivery system. For instance, hyaluronan-conjugated core-shell lipid nanoparticles comprising of chitosan core and lipid shell are used to target CD44 positive retinal pigment epithelium cells via intravitreal injection, and core-shell polymer microspheres with PLGA core and poly(L-lactic acid) (PLLA) shell encapsulate the brimonidine tartrate to achieve sustained IOP reduction in the treatment of glaucoma by subconjunctival implantation [25, 26]. However, there are still no studies on the core-shell type LPNs consisting of PLGA core and lipid shell using to treat eye diseases via topical administration such as eyedrops. As a result, LPNs which consist of lipid shell and PLGA core hold great promise for topical ocular drug delivery.
Brinzolamide (Brz), a second-generation carbonate anhydrase inhibitor, has been widely used in the treatment of glaucoma and intraocular high-pressure due to its outstanding effect in IOP reduction. However, due to its poor solubility in water at pH 7.4 (25 ℃), the commercial available formulation of Brz (AZOPT®) is a suspension for its poor ocular bioavailability and patient compli-ance in clinical practice [27]. With the aim to enhance the solubility, improve the ocular bioavailability and provide high patient compliance of Brz, Brz was loaded by nanocarriers previously by our group [28, 29]. Liposomes offered efficient loading of Brz with simple methods. Moreover, it showed good biocompatibility with no eye irritation. Disappointingly, Brz liposomes had a limited to corneal permeability and duration in intraocular pressure reduction which resulted in less effective in improving bioavailability. In order to enhance bioavailability via increasing corneal permeability and achieving sustained release of Brz, a novel and integrated system known as LPNs could be used for topical ocular drug delivery.
In this study, we combined PLGA nanocore and lipid shell to formulate Brz-LPNs. It is worth noting that LPNs consisting of PLGA core and soybean phosphatidylcholine (SPC) shell are used for topical ocular delivery for the first time. Brz-LPNs were optimized in terms of mean particle size and encapsulation efficiency. The pharmaceutical properties of optimal Brz-LPNs were then charac-terized systemically including particle size, zeta potential, entrapment efficiency (EE) of Brz. Transmission electron micro-scope (TEM) and fourier transformed infrared spectra (FTIR), stability and in vitro release profile of ptimal Brz-LPNs were further studied. Moreover, ex vivo corneal permeation, cytotoxicity and in vivo IOP reduction efficiency of Brz-LPNs were also evaluated.
According to the literature, Brz-LPNs were prepared by the two-step approach with some modifications [30]. In short, the lipid shell and the polymeric core were prepared individually before being fused together by direct hydration. First, soybean phosphatidylcholine (SPC) and cholesterol were completely dissolved in mixed solvent with chloroform/methanol (4:1, v/v). Subsequently, the organic solvents were got rid of by rotary evaporator and meanwhile a thin film of lipid was produced at 37 ℃. Brz-NPs were fabricated by emulsion solvent evaporation method [31]. Specifically, Brz and PLGA were dissolved in acetone and dichloromethane. Subsequently, the organic phase was added into PVA solution and then the emulsion (O/W) was obtained by probe sonication in ice bath. The organic solvents were rapidly removed by evaporation under vacuum at 37 ℃ to obtain Brz-NPs. Afterwards, Bra-NPs prepared above were added into preformed lipid film. The hydration was conducted at 60 ℃ to promote restructuring of the lipid onto Brz-NPs surface. To obtain Brz-LPNs, the acquired suspension was sonicated for 3 min at 100 W in an ice bath and filtered through a 0.22-mm syringe filter for purpose of sterilization. The blank lipid nanoparticles (blank-LPNs) were prepared following the same method without the addition of Brz.
The main characteristics of Brz-LPNs were measured by a Zetasizer (Zetasizer Nano-ZS 90; Malvern Instruments Ltd., Malvern, UK) at 25 ℃, including the mean particle size, size distribution and zeta potential of Brz-LPNs. The prepared Brz-LPNs were suitably diluted with deionized water and experiments were conducted in triplicate. The encapsulation efficiency (EE) of Brz-LPNs was determined by high performance liquid chromatography (HPLC, Waters Alliance 2695) as previously described [32]. EE was calculated by the following calculation equation: EE = (initial Brz amount -the amount of Brz in supernatant)/initial Brz amount 100%. Analyses were performed in triplicate and the values were expressed as mean± SD.
The morphology characterization of Brz-LPNs was examined by TEM (TEM, H-600, Hitachi, Japan). After dilution with deionized water, the Brz-LPNs samples were placed on copper electron microscopy grids and were negatively stained with 2% (w/v) phosphotungstic acid solution for inspection at an acceleration voltage of 100 kV.
FTIR spectroscopy was obtained using Bruker Vector 22 FT-IR Spectrometer (Bruker, Switzerland) to reveal whether Brz was successfully encapsulated into LPNs. Spectra of pure Brz, physical mixture of Brz and blank-LPNs (PM), blank-LPNs, and Brz-LPNs were obtained using the KBr pellet method. Each KBr disk was scanned at 4 mm/s over a wave number region of 400–4000 cm-1. Brz-LPNs were stored at 4 ℃ for 15 days to inspect the preliminary stability. The changes in particle size and EE of Brz-LPNs were tested by the methods as described above.
To further investigate the drug release of Brz-LPNs. The in vitro release profile of Brz from Brz-LPNs was investigated in simulated tear fluid (STF) by dynamic dialysis technique [33]. In brief, free Brz, Brz-LPs and Brz-LPNs were put in dialysis bags (MWCO 144, 000 Da) and were shaken at 37 ℃ with a speed of 100 rpm, respectively. At a certain time interval, 0.5 mL of the release medium was removed and replaced with 0.5 mL of the fresh release medium. The content of Brz was tested by HPLC as described above. Analyses were performed in triplicate.
Corneal permeability of ophthalmic preparations is important. Therefore, the corneal permeability of Brz-LPNs was investigated by the Franz diffusion chamber consisting of a donor and a receiver compartment (with a volume of 0.6 and 1.0 mL, respectively). The diffusion chamber was maintained at a constant temperature (37 ±0.2 ℃) with mixing conditions using a magnetic stirrer. The cornea together with a 2 mm ring of sclera was excised within 20 min after the rabbits were sacrificed and stored in glutathione-bicarbonate-Ringer buffer before use. The gained cornea was firstly fixed between clamped donor and receptor compartments through the sclera. 1 mL of the GBR solution was added to the endothelial side, while 0.5 mL of AZOPT®, Brz-LPs or Brz-LPNs was added to the epithelial side. Next, 0.3 mL of samples were withdrawn at 15, 30, 60, 90, 120, 180, 240, 300, and 360 min and replaced with fresh glutathione-bicarbonate-Ringer solution. The amount of drug permeation across the cornea was estimated by the HPLC method as described above. The experiment was done in triplicate.
To observe the safety of Brz-LPNs. Cytotoxicity assay of Brz-LPNs was carried out by MTT assay on L929 cells [34]. Briefly, L929 cells were seeded into 96-well plate at an initial density of 3×103 cells per well in 100 mL medium and cultivated for 24 h at 37 ℃. Cells were then treated with various concentrations of blank-LPNs or Brz-LPNs for 48 h. Finally, cytotoxicity of Brz-LPNs was evaluated by MTT assay as previously described [35].
IOP was recorded as previously described [36]. Fifteen rabbits were divided into three groups. AZOPT®, blank-LPNs and Brz-LPNs were applied with the volume of 50 mL and IOP was measured at a certain time, respectively. In the study, the right eye of all rabbits was instilled topically into the upper quadrant and the left eye of all rabbits was used as a control without treatment. IOP was surveyed at specific time 0.5, 1, 2, 4, 8, 12, 16, 20 and 24 h using Schiotz Tonometer (Rudolf Riester GmbH and Co., KG, Germany) after formulations administration. In addition, for the histopathological analysis, the treated cornea was immersed in a 4% paraformalde-hyde solution. Then, the fixed tissue was embedded in paraffin and sectioned at 5 μm, and processed for hematoxylin and eosin (H & E) staining. Images were acquired on a light microscope (Olympus, Tokyo, Japan).
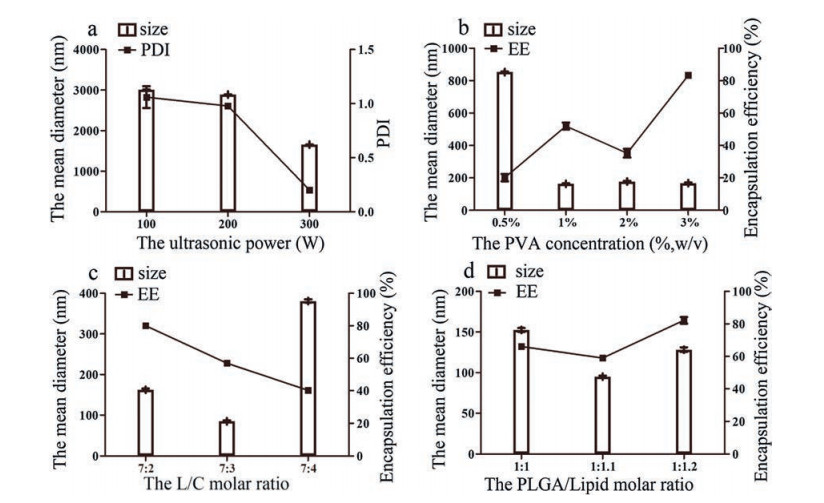
According to the literature, the parameters, such as particle size, PDI and drug EE, are critical properties for nanocarriers because of the influence on stability, drug release profile, drug penetration, and so on [37-39]. Hence in order to achieve a low mean particle size and high drug EE, the technological parameters of Brz-LPNs were systemically optimized on account of affecting the property of formulations, including the ultrasonic power, the concentration of PVA, the lipid-to-cholesterol (L/C) molar ratio and the PLGA-to-lipid molar ratio.
The size of formulations was directly affected by the ultrasonic power [40]. The higher ultrasonic power would generate smaller particle size and PDI due to the high net shear stress. Therefore, it was clear that remarkable particle size and PDI reduction was noticed along with the increase of ultrasonic power. Therefore, the influences of the ultrasonic power on particle size and the polymer dispersity index (PDI) were investigated. As shown in Fig. 1a, the particle size and PDI were successively decreased with the ultrasonic power increase. The ultrasonic power led to smaller mean diameter and narrower PDI when it was 300 W. As a result, the favorable ultrasonic power was identified 300 W. On the basis of previous studies, PVA was generally used as a stabilizer in the preparation of PLGA with a certain concentration of PVA [41]. The mechanism of PVA binding was the penetration of PVA and PLGA molecules during the formulations preparation [42]. In this study, the influence of the concentration of PVA was necessary to be evaluated. In Fig. 1b, the particle size and the EE of lipid nanoparticles were distinctly affected by the PVA within the concentration range of 0.5% (w/v) to 3% (w/v). The particle size first decreased sharply and then gradually stabilized when the concentration of PVA increased. Whereas, the EE of Brz-LPNs exhibited a fluctuant tendency as PVA concentration increased. Brz-LPNs could be provided with smaller particle size and higher EE when PVA concentration was 3% (w/v). According to the results, the particle size decreased probably with the concentration of PVA increased. The reason is that the excess amount of PVA residing at organic solvent/aqueous interface results in a reduced surface tension [43]. As is well-known, cholesterol has an effect on particle size and EE of lipid formulations because cholesterol had an influence on the ordered arrangement and the flexibility of the lipid membrane in the preparation process [44]. Thereby the lipid-to-cholesterol (L/C) molar ratio was optimized. As seen in Fig. 1c, the mean diameter firstly decreased and then increased dramati-cally with the increase of cholesterol molar percent. In contrast, the EE was significantly reduced by cholesterol molar percent increasing. The reason was that increased cholesterol proportion might cause the decreased extent of encapsulation of the drug [45]. When the lipid-to-cholesterol (L/C) molar ratio was fixed to 7:2, both the particle size and the EE were preferable. Furthermore, the proportion of lipid to polymer was regarded as an important parameter affecting particle size and EE of lipid nanoparticles [46]. In order to obtain the best Brz-LPNs, the molar ratio of PLGA-to-lipid (PLGA/lipid) was also optimized. As shown in Fig. 1d, the formulations obtained better size and higher EE when the PLGA-to-lipid molar ratio was 1:1.2. The probable reason might be the increased affinity between formulations and hydrophobic drug with increased polymer proportion [47]. Based on the above optimized conditions, the final formulation of Brz-LPNs were successfully prepared with 300 w ultrasonic power, 3% (w/v) PVA concentration, 7:2 molar ratio of L/C and 1:1.2 molar ratio of PLGA/Lipid.
Figure 1
Figure 1.
Effect of various processing parameters on the particle size (nm), the polymer dispersity index (PDI) and encapsulation efficiency (EE) of Brz-LPNs, including (a) the ultrasonic power (W), (b) the concentration of PVA (%, w/v), (c) the lipid-to-cholesterol (L/C) molar ratio and (d) the PLGA-to-lipid (PLGA/Lipid) molar ratio. Mean ±SD (n = 3).
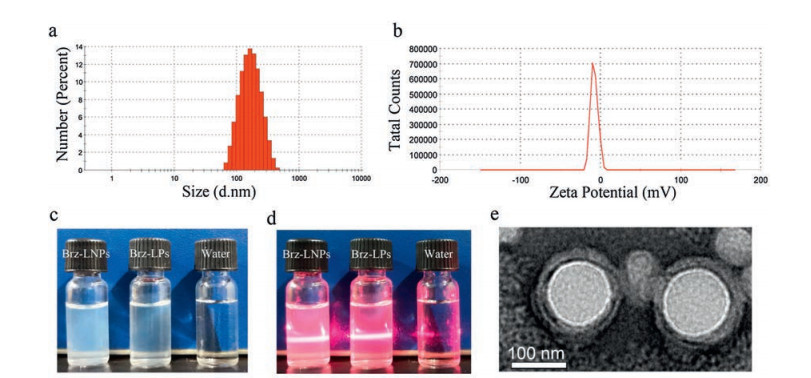
The optimal Brz-LPNs obtained a high EE of 86.7% 2.28%. The average particle size was 151.23 1.64 nm (Fig. 2a) with narrow size distribution. The zeta potential of Brz-LPNs was slightly negative, with the value of 7.53 0.52 mV (Fig. 2b). To investigate the surface morphology of our optimal formulations, TEM analysis was performed. As shown in Fig. 2e, TEM image of Brz-LPNs was confirmed that the nanocore was generally spherical in shape and was well coated by bilayer lipid [48]. The colloidal solution was observed as slightly blue opalescence with strong Tyndall effect compared with water (Figs. 2c and d).
Figure 2
Figure 2.
Characterization of the optimal Brz-LPNs. (a) Size distribution; (b) Zeta potential; (c) The appearance and (d) Tyndall effect of Brz-LPNs; (e) TEM image of Brz-LPNs.
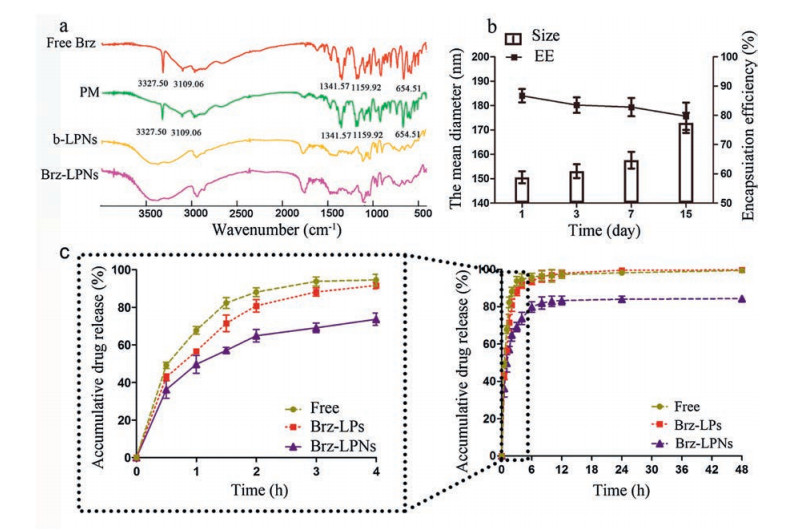
The FTIR analysis was carried out to confirm that Brz was absolutely and successfully encapsulated into Brz-LPNs. Hence, the results of successful encapsulation of Brz by LPNs were evaluated by FTIR. FTIR spectra of free Brz, PM, blank-LPNs and Brz-LPNs were showed in Fig. 3a. According to the characteristic peaks of free Brz,it noticed that the intensity of S=O stretch (1341.57 and 1159.92 cm-1), C-NH-C stretch (3327.50 cm-1), CH3 stretch (2975.35 cm-1), and CH2 stretch (2852.62 and 1456.18 cm-1). In addition, the absorption peaks at 3109.06 and 654.51 cm-1 attributed to the existence of heterocycle including N and S elements [49]. The peaks of Brz at 3327.50, 3109.06, 1341.57, 1159.52, and 654.51 cm-1 were distinct from Brz-LPNs with a clear loss in the intensity and resolution of peaks. FTIR spectra of different formulations displayed that the characteristic peaks of Brz disappeared completely from the infrared absorption spectra of Brz-LPNs, indicating that Brz was absolutely and successfully encapsulated into Brz-LPNs [28, 36].
Figure 3
Figure 3.
Characterization of Brz-LPNs. (a) FTIR spectra of free Brz, physical mixture of Brz and blank-LPNs (PM), blank-LPNs (b-LPNs) and Brz-LPNs; (b) Change in size and EE of Brz-LNPs in 2 weeks; (c) In vitro release kinetics of free Brz, Brz-LPs and Brz-LPNs in simulated tear fluid. Mean ± SD (n = 3).
Brz-LPNs displayed a favorable stability with no detectable changes in particle sizes and EE at least one week in 4 ℃ (Fig. 3b). However, when Brz-LPNs were stored over two weeks, an obvious enlargement of particle size and a reduction of EE of Brz were observed. In summary, Brz-LPNs were suggested to be stable in 4 ℃ at least one week. To improve instability issues of Brz-LPNs, other effective strategies could be used such as lyophilization or other stabilization techniques [50].
In vitro release profile of Brz from Brz-LPNs was illustrated in Fig. 3c. About 94.5%, 91.5% and 72.4% Brz were released from free Brz, Brz-LPs and Brz-LPNs within a period of 4 h, respectively. After 12 h, there almost 100% Brz were released from free Brz and Brz-LPs, respectively, but merely 83.2% Brz was released from Brz-LPNs. Hence, the in vitro release study indicated that Brz-LPNs appeared a slower sustained release compared to free Brz and Brz-LPs. The possible reason was that the architecture of Brz-LPNs was polymeric core encircled by phospholipids layers that can regulate drug release profiles [51]. According to the result, the release of Brz out of Brz-LPNs was slower than Brz-LPs and AZOPT®, which might be attributed to the fact that their unique core-shell structure could lead to tunable and sustained drug release profiles [52]. The release of drugs from the system required overcoming two barriers. The first barrier was polymeric core composed of a biodegradable hydrophobic polymer. The polymeric core could encapsulate poorly water-soluble drugs and control drug release from the system. The second barrier is the shell composed of lipid layers, and the shell could be conducive to reduce drug diffusion from the core [53]. Therefore, Brz-LPNs had the ability to increase drug encapsulation and alter drug release rates.
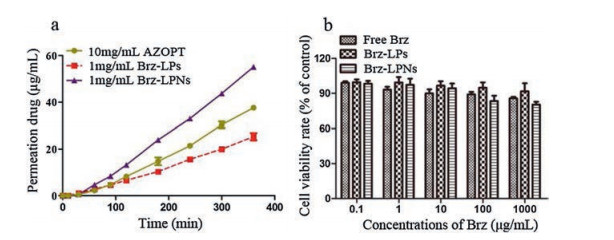
As is known to us, it is a challenging task for an ocular drug to improve the bioavailability of drugs administered topically. One of approaches to increase ocular drug bioavailability is enhancing penetration of the drug [54]. Hence, the main objective was to evaluate the cumulative amount of drug permeated the cornea. The cumulative amount of Brz (mg) of AZOPT®, Brz-LPs and Brz-LPNs in the receptor medium at the time range 0–360 min was shown in Fig. 4a. There was no significant difference between Brz-LPs and AZOPT® within a period of 60 min. However, there was an obvious distinction appeared at 120–360 min. Brz-LPNs showed a significant increase of Brz cumulative permeation compared with Brz-LPs and AZOPT®. The cytotoxicity assays of free Brz, Brz-LPs and Brz-LPNs were carried out with L929. As shown in Fig. 4b, the MTT assay indicated that almost 90% of L929 cell viability with free Brz, Brz-LPs and Brz-LPNs at the concentration range 0–1000 μg/mL. Therefore, there was almost no toxicity to the cells by Brz-LPs and Brz-LPNs in the drug concentration range 0–1000 μg/mL. According to our results, it was found that the quantities of permeation drug were significantly higher with the application of Brz-LPNs than Brz-LPs and AZOPT®. The favorable penetration of Brz-LPNs across the cornea could be attributed to the architecture of LPNs which was polymeric core encircled by lipids layers so that it could realize high encapsulation of hydrophobic drugs and slowly delivery drug to the precorneal area [55]. The cytotoxicity assays of Brz-LPNs proved that final formulations had almost no toxicity to the cells in the drug concentration range 0–1000 μg/mL.
Figure 4
Figure 4.In vitro corneal permeability studies and cytotoxicity assays of Brz-LPNs. (a) In vitro corneal permeability of Brz commercial formulation (AZOPT®), Brz-LPs and Brz-LPNs; (b) The cytotoxicity assays of Free Brz, Brz-LPs and Brz-LPNs. Mean ± SD (n = 3).
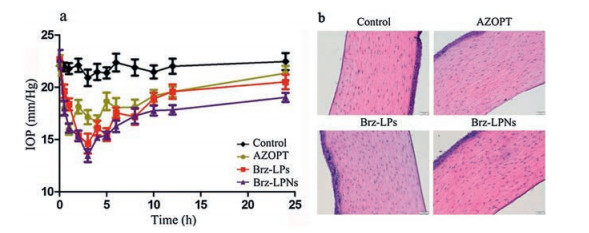
The IOP reduction was observed after the administration of the formulations (AZOPT®, Brz-LPs and Brz-LPNs). According to the literature, 15% or more IOP reduction was defined as an effective therapy in IOP control [56]. The IOP-lowering effect of Brz-LPNs was examined following one topical eyedrop administration by comparison with AZOPT® and Brz-LPs. As shown in Fig. 5a, the trend of IOP-lowering effect with different Brz formulations was all had effect of lowering intraocular pressure after administration. Compared to the normal IOP, AZOPT® was observed that achieved a maximum IOP reduction of 26.71% after 1 h. On the contrary, it was clear that the maximum IOP reduction of 30.44% for Brz-LPs formulations and 39.31% for Brz-LPNs formulations were detected after 3 h, respectively. However, it is worth noting that AZOPT® and Brz-LPs had a limitation to effectively reduce IOP in the dosed eyes at 10 h and subsequent time points. Brz-LPNs displayed sustained and effective IOP reduction over a period of 24 h. As compared to AZOPT® and Brz-LPs, Brz-LPNs exhibited an enhanced and sustained IOP reduction was within a period of 24 h. The enhanced IOP reduction of Brz-LPNs might be attributed to the presence of phospholipid as an additional layer facilitating the binding of Brz-LPNs with corneal membrane and probably extending the absorption of Brz [57]. In addition, the sustained IOP reduction of Brz-LPNs might be attributed to the presence of lipid as a coating layer on nanoparticles, as well as attributed to nano-size of Brz-LPNs [48].
Figure 5
Figure 5.
(a) The IOP reduction observed after administration of Brz in AZOPT®, Brz-LPs and Brz-LPNs. Data were compare with respect to control eyes of each formulations. Mean ± SD (n = 5). (b) Representative H & E images (200X) of the cornea of New Zealand rabbits treated with the saline (Control), AZOPT®, Brz-LPs and Brz-LPNs.
It is necessary to develop an effective and safety formulation for ocular drug delivery. The results of corneal pathology analysis using a light microscope were shown in Fig. 5b. After treatment with AZOPT®, Brz-LPs and Brz-LPNs, the corneal epithelium cells of the rabbits were intact. Besides, there was no obvious inflamma-tory cell invasion and proliferation in corneal stromal cells of each preparation, indicating that there was no edema and inflammatory reaction in the cornea. Compared with the saline group, the Brz-LPs and Brz-LPNs groups showed no significant difference in corneal pathology, indicating that the preparation had no obvious toxic effects on rabbit cornea.
In this work, Brz-LPNs, as novel core-shell type of ocular drug delivery systems, were successfully developed and were system-atically optimized. The optimal Brz-LPNs obtained a higher EE and an ideal particle size with a narrow size distribution. The core-shell structure of Brz-LPNs was confirmed that the nanoparticles were generally spherical in shape and were well surrounded by bilayer lipid. Brz-LPNs displayed good storage stability and a better sustained release performance than Brz-LPs. It is satisfied that ex vivo corneal permeation of Brz-LPNs was significantly increased compared with Brz-LPs and AZOPT®. According to research literature, the corneal penetration of ocular drugs is related to improve drug bioavailability [58]. Hence, it is meaningful to enhance ex vivo the corneal penetration of ocular drugs. In vivo experiment showed that Brz-LPNs (1 μg/mL Brz) achieved a more sustained and effective reduction in IOP than AZOPT® (10 μg/mL Brz) and Brz-LPs in white New Zealand rabbits, which signified that Brz-LPNs had a sustained and enhanced therapeutic effect compared AZOPT® and Brz-LPs. In summary, all these data supported the belief that Brz-LPNs would have a promising future as a novel formulation of Brz for glaucoma treatment.
Acknowledgments
This work was financially supported by Sichuan Province Science and Technology Support Program (Nos. 16ZC2698 and 2018JY0582) and the National Natural Science Foundation of China (No. 81872821).
Figure 1
Effect of various processing parameters on the particle size (nm), the polymer dispersity index (PDI) and encapsulation efficiency (EE) of Brz-LPNs, including (a) the ultrasonic power (W), (b) the concentration of PVA (%, w/v), (c) the lipid-to-cholesterol (L/C) molar ratio and (d) the PLGA-to-lipid (PLGA/Lipid) molar ratio. Mean ±SD (n = 3).
Figure 2
Characterization of the optimal Brz-LPNs. (a) Size distribution; (b) Zeta potential; (c) The appearance and (d) Tyndall effect of Brz-LPNs; (e) TEM image of Brz-LPNs.
Figure 3
Characterization of Brz-LPNs. (a) FTIR spectra of free Brz, physical mixture of Brz and blank-LPNs (PM), blank-LPNs (b-LPNs) and Brz-LPNs; (b) Change in size and EE of Brz-LNPs in 2 weeks; (c) In vitro release kinetics of free Brz, Brz-LPs and Brz-LPNs in simulated tear fluid. Mean ± SD (n = 3).
Figure 4In vitro corneal permeability studies and cytotoxicity assays of Brz-LPNs. (a) In vitro corneal permeability of Brz commercial formulation (AZOPT®), Brz-LPs and Brz-LPNs; (b) The cytotoxicity assays of Free Brz, Brz-LPs and Brz-LPNs. Mean ± SD (n = 3).
Figure 5
(a) The IOP reduction observed after administration of Brz in AZOPT®, Brz-LPs and Brz-LPNs. Data were compare with respect to control eyes of each formulations. Mean ± SD (n = 5). (b) Representative H & E images (200X) of the cornea of New Zealand rabbits treated with the saline (Control), AZOPT®, Brz-LPs and Brz-LPNs.

 DownLoad:
DownLoad:

 下载:
下载:






