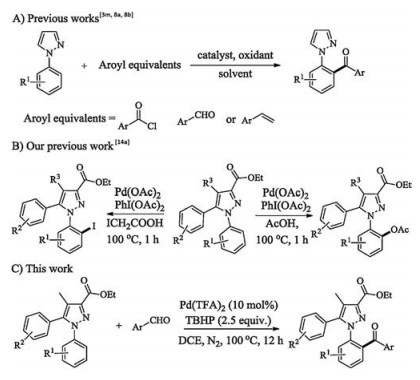
Scheme 1.
Previous related works and our present work.
Palladium-catalyzed late-stage mono-aroylation of the fully substituted pyrazoles via aromatic C-H bond activation
Miao-Miao Chen , Ling-Yan Shao , Li-Jun Lun , Yu-Liang Wu , Xiao-Pan Fu , Ya-Fei Ji

Diaryl ketones are prevalent structural motifs broadly distributed in pharmaceuticals, fragrances, agrochemicals, dyes, organic functional materials, food additives, and natural products [1]. The electrophilic Friedel–Crafts aroylation of arenes as the most classical access to the architechtures usually needs large amounts of Lewis acids or strong mineral acids, which is extremely limited by electronically biased substrate, poor regioselectivity, narrow functional group tolerance and stoichiometric waste [2]. Recently, transition-metal-catalyzed aroylation assisted by auxiliary has emerged as a powerful tool to circumvent the limitations. Considering atom- and step-economy, many colleagues have successfully developed a series of direct aroylations by using aldehydes [3], alcohols [4], α-oxocarboxylic acids [5], α-diketones [6a], carboxylic acids [6b], anhydrides [6c], amines [6d], benzylic ethers [6e], toluene derivatives [7] and others [8] as aroyl sources with the assistance of various directing groups. Therein, the excellent directing ability of pyrazole moiety for aroylation of arenes have been uncovered by Frost's and other following works (Scheme 1A) [3m, 8a, 8b].
Currently, late-stage functionalization, which conducts a direct transformation in lead or privileged structures for generating greatly potential drug-like molecules or highly valuable scaffolds [9], has turned into a leading strategy in chemistry community. As a result, many elegant efforts have been devoted to a variety of late stage diversification of bioactive compounds [10], drug leads [11] and molecular materials [12], demonstrating supernormal value of this strategy.
Given that the highly functionalized pyrazoles and thiazoles serve as important pharmacophores in drug discovery [13], We recently reported efficient late-stage mono-acetoxylation/iodin ation of fully substituted pyrazoles (Scheme 1B), as well as late stage mono-acyloxylation of the highly substituted thiazoles [14]. Albeit with the advances, late-stage functionalization directed by inherent pharmacophore still remains to be developed. To continue our interest, herein, we disclosed an efficient protocol for the regioselective late-stage mono-aroylation of multifunction al pyrazoles using aldehydes as aroyl surrogates, through a Pd catalyzed aromatic C—H activation with aroyl radical coupling (Scheme 1C).
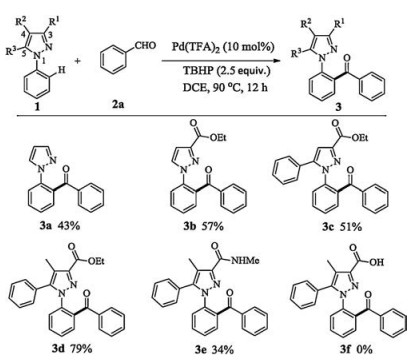
Our initial effort focused on briefly evaluating the substituted pattern of pyrazole directing group. To our delight, the unsub stituted 1-phenyl-1H-pyrazole 1a with benzaldehyde (2a, 2.0 equiv.) delivered the expected product 3a in 43% yield in the presence of Pd(TFA)2 (10 mol%) and TBHP (2.5 equiv.) in DCE (2.0 mL) under N2 atmosphere at 90 ℃ for 12 h (Fig. 1). When an ethoxycarbonyl group was installed at 3-position, the yield was evidently increased to 57%, implying possible synergic role of the group (3b). Sequentially, incorporating phenyl into 5-position led to a slight drop with 51% yield (3c). In striking contrast to the substrates (1a-c), the fully substituted pyrazole dramatically furnished a good yield of 79% (3d). However, the yield was drastically diminished to 34% upon changing ethoxycarbonyl into carbamyl at 3-position (3e). Even more, no any reaction occurred when the ethoxycarbonyl was varied as a carboxyl (3f). Thus, the fully substituted 1d was selected as an optimal directing group to effectively conduct the desired aroylation.
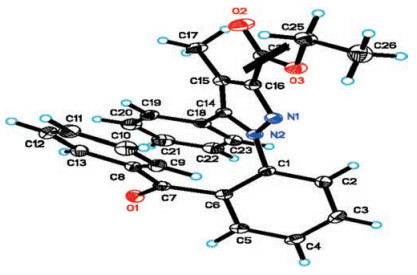
Upon the directing group ensured, we turned our attention to investigating main reaction parameters with the model substrates 1d and 2a (Table 1). Firstly, screening of Pd salts, including Pd (TFA)2, Pd(OAc)2, Pd(PPh3)2Cl2, PdCl2 and Pd(CH3CN)2Cl2, exhibited Pd(TFA)2 holding the highest catalytic activity (entries 1 —5). Whereas lowering the loading of Pd(TFA)2 to 5 mol% obviously cut down the yield (entry 1 versus 6). Subsequently, it was found that DCE was the best solvent compared to other common ones (entry 1 versus 7–10). Further testing peroxides, organic and inorganic oxidants such as DCP, TBPB, PhI(OAc)2, K2S2O8 and BQ, indicated that TBHP was the ideal one (entry 1 versus 11 —15). Furthermore, the yield was impaired when decreasing the stoichiometry of TBHP to 2.0 equiv. (entry 1 versus 16). On the other hand, raising the stoichiometry of TBHP to 3.0 equiv only gave rise to a similar yield (entry 1 versus 17). Considering the impact of the reaction temperature, it was observed that the yield slightly increased to 83% under 100 ℃ (entry 18). But further elevating the temperature to 110 ℃ resulted in a drop of yield (entry 19). Besides, the N2 protection proved to be vital for the transformation (entry 20). Hence, we chose entry 18 as the standard reaction conditions for the following aroylation survey. At last, the geometrical configuration of 3d was confirmed by X-ray single crystal crystallography (Fig. 2, details see Supporting information). The crystal structural analysis exhibits that the central pyrazole ring do not share a common plane with 1, 5-diphenyl, consequently, avoiding potential secondary aroylation [14a].
With the optimized reaction conditions in hand, we commenced to evaluate the scope of functionalized pyrazoles (Fig. 3). The substrates with para-electron-donating and electron-withdrawing groups at 1-phenyl could favourably afford the desired products (3g—l). Therein, the substrate withpara-OMe gracefully supplied an excellent yield of 87% (3g). While the substrate containing strongly electron-withdrawing and coordinating NO2 [3j, 7b, 15] was successfully transformed into the aroylated product despite in 39% yield (3l). Generally, the aroylations of meta-substituted substrates appropriately took place at the sterically yield of 71%, demonstrating the reaction compatible with fused arene. With 5-aryl further tested, the substrates with an electrondonating group (R2 =para-OMe) and an electron-withdrawing group (R2 =para-Cl) also underwent the protocol providing good yields of 80% and 78%, respectively (3w and 3x). Furthermore, a respected yield of 43% was observed for the substrate containing 5- thiophenyl (3y). Besides, the aroylation via six-membered activation mode also worked well to furnish 63% yield (3z). A lower yield of 69% was observed when the methoxycarbonyl instead of ethoxycarbonyl at 3-position (3A versus 3d). Lastly, installing ethyl and isopropyl at 5-position only provided 46% and 36% yields, respectively, demonstrating that aryl group at 5-position is crucial for the transformation (3B and 3C).
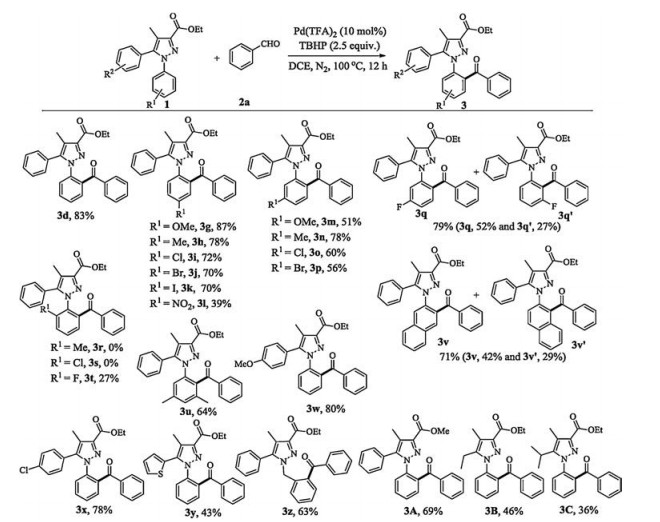
Next, various arylaldehydes were investigated for the aroylation (Fig. 4). It is worth noting that the ortho-hindered aromatic aldehydes were feasible coupling partners with 63%–88% yields, not to show obvious electronic effect (4a—d). Similarly, the meta less hindered ortho-positions (3m-p). Interestingly, as for the meta-F substituted substrate, the aroylation occurred at both ortho-positions, respectively, in an overall yield of 79% (3q and 3q'). Nicely, the reactive Cl, Br, I, NO2 and innate ethoxycarbonyl groups are readily elaborated for further synthetic transformation.
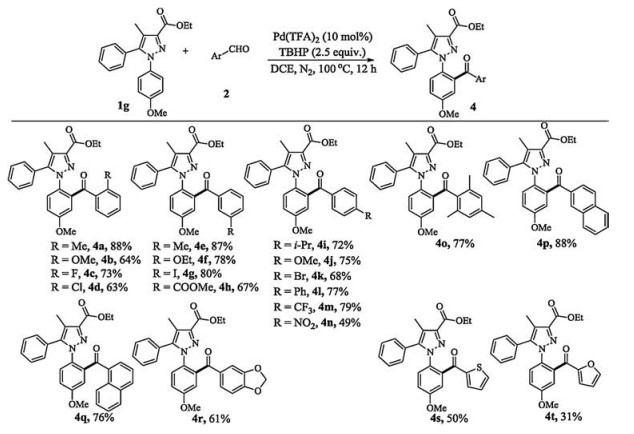
We paid a special attention to the substrates containing ortho-substituted 1-aryl. Usually, no any aroylated product was detected because of the highly twisted molecular configuration (3r and 3s) [14a]. Impressively, with respect to much less bulk of F, the substrate 1t afforded a synthetically useful yield of 27% (3t). Remarkably, the substrate with a crowded meta, meta '-disubstituted moiety also provided a moderate yield of 64% (3u). The 1-(2- nathphyl)-substituted pyrazole was amenable to the protocol giving the major product 3v and minor product 3v' in an overall yield of 71%, demonstrating the reaction compatible with fused arene. With 5-aryl further tested, the substrates with an electrondonating group (R2 = para-OMe) and an electron-withdrawing group (R2 =para-Cl) also underwent the protocol providing good yields of 80% and 78%, respectively (3w and 3x). Furthermore, a respected yield of 43% was observed for the substrate containing 5- thiophenyl (3y). Besides, the aroylation via six-membered activation mode also worked well to furnish 63% yield (3z). A lower yield of 69% was observed when the methoxycarbonyl instead of ethoxycarbonyl at 3-position (3A versus 3d). Lastly, installing ethyl and isopropyl at 5-position only provided 46% and 36% yields, respectively, demonstrating that aryl group at 5-position is crucial for the transformation (3B and 3C). Next, various arylaldehydes were investigated for the aroylation (Fig. 4). It is worth noting that the ortho-hindered aromatic aldehydes were feasible coupling partners with 63%–88% yields, not to show obvious electronic effect (4a—d). Similarly, the metaandpara-substituted arylaldehydes suffered from the protocol offering good to excellent yields (4e—m). Nevertheless, the strongly coordinating NO2 [3j, 3j, 7b, 3j, 15] substituted benzaldehyde was less reactive, possibly due to lower catalytic overturn (4n). Notably, the highly hindered methyl trisubstituted substrate could proceed favourably with 77% yield (4o). Moreover, 2-naphthylaldehyde and 1-naphthylaldehyde were greatly suitable substrates to afford good to excellent yields (4p and q). Besides, electron-rich piperonal was converted to the corresponding product in moderate yield (4r). Furthermore, the heterocyclic aldehydes also participated in the protocol providing the aroylated products in synthetically useful to moderate yields (4s and 4t). However, our additional investigation indicated that aliphatic aldehydes such as propionaldehyde and phenylacetaldehyde did not take effect under the work conditions. It should be noted that the reaction only happened at single ortho-position to exclusively afford the mono-aroylation products in all cases, due to the twisted configurations.
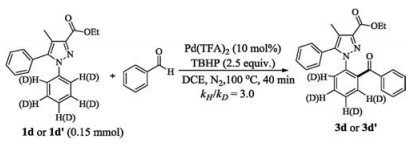
To gain insight into the reaction mechanism, the reaction between 1d and 2a was carried out in the presence of 2.0 equiv. TEMPO [(2, 2, 6, 6-tetramethylpiperidin-1-yl)oxy] as a radical scavenger. The TEMPO ester 5a was isolated in 63% yield with no aroylated product (3d) observed, suggesting the aroyl radical generated in the protocol (Scheme 2) [16]. In addition, an intermolecular competition reaction between 1d and 1d' (1d-d5) with 2a was conducted in one vessel exhibited a notable primary kinetic isotopic effect (kH/kD = 3.0, see Supporting information). The result implies that the Csp2–H bond cleavage might be involved in the rate-limiting step (Scheme 3).

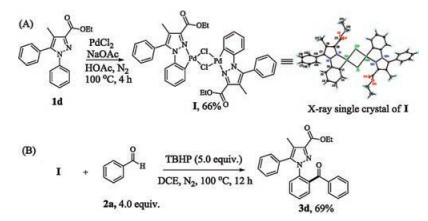
Gladly, we successfully prepared a crystallizable palladacycle I according to the literature [17]. Its dual-core dimeric structure was unambiguously confirmed by X-ray single crystal crystallography (Scheme 4A, details see Supporting information). Omitting Pd (TFA)2 and 1d, the dimer I smoothly converted into the target product 3d in 69% yield under other identical conditions (Scheme 4B, details see Supporting information). Therefore, the dimer I was considered to be a key palladacycle in the process.
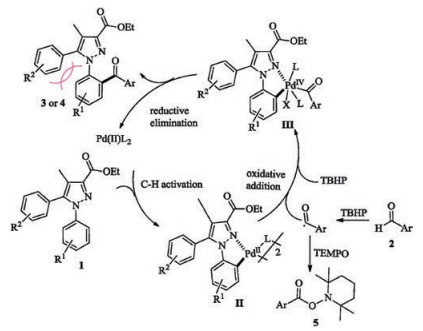
On the basis of the experimental results and previous reports [3a, b, g, h, j, k, l], a plausible reaction mechanism is proposed in Scheme 5. Firstly, the substrate 1 coordinates with Pd(II) catalyst followed by a Csp2–H bond activation to form a dual-core dimeric cyclopalladated intermediate II. Then, the dimer II undergoes an oxidative addition with aroyl radical, generated in situ by a hydrogen abstraction, to produce the highly active Pd(IV) species III. In the process, Pd(II) and the radical conduct a single electron transfer (SET), followed by an oxidation of Pd(III) into Pd(IV) with TBHP [18]. Herein, TBHP would play the roles of both radical initiator and oxidant [3c, 7a]. Finally, the adduct III is subjected to a following reductive elimination to release the aroylated product 3 or 4, and regenerate the Pd(II) catalyst.
In summary, we have developed a Pd-catalyzed late-stage aroylation of 4-methyl-1, 5-diaryl-1H-pyrazole-3-carboxylates via direct Csp2–H bond activation with broad substrate scope and good functional group tolerance. The protocol is featured by the highly functionalized substrates, exclusive mono-site-selectivity and simple operation. Particularly, a dual-core dimeric palladacycle is confirmed by X-ray crystallography to profitably reveal a plausible reaction mechanism. It is expected that the protocol can serve as a potential tool for further elaboration from pharmaceutically important and highly functionalized pyrazoles.
We gratefully thank the National Natural Science Foundation of China (Nos. 21476074 and 21676088) for financial support.
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.09.022.
(a) P.J. Masson, D. Coup, J. Millet, N.L. Brown, J. Biol. Chem. 270 (1995) 2662-2668;
(b) P.J. Harrington, E. Lodewijk, Org. Process Res. Dev. 1 (1997) 72-76;
(c) S. Gmouh, H.L. Yang, M. Vaultier, Org. Lett. 5 (2003) 2219-2222;
(d) K.R. Romines, G.A. Freeman, L.T. Schaller, et al., J. Med. Chem. 49 (2006) 727-739;
(e) N.A.McGrath, M.Brichacek, J.T.Njardarson, J.Chem.Educ.87 (2010)1348-1349. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM7852334
(a) I. Hachiya, M. Moriwaki, S. Kobayashi, Tetrahedron Lett. 36 (1995) 409-412;
(b) I.V. Kozhevnikov, Appl. Catal. A-Gen. 256 (2003) 3-18;
(c) G. Sartori, R. Maggi, Chem. Rev. 106 (2006) 1077-1104. http://www.sciencedirect.com/science/article/pii/004040399402221V
(a) X.F. Jia, S.H.Zhang, W.H.Wang, F. Luo, J.Cheng, Org. Lett.11 (2009)3120-3123;
(b) C.W. Chan, Z.Y. Zhou, A.S.C. Chan, W.Y. Yu, Org. Lett. 12 (2010) 3926-3929;
(c) O. Basle', J. Bidange, Q. Shuai, C.J. Li, Adv. Synth. Catal. 352 (2010) 1145-1149;
(d) Y.N. Wu, B.Z. Li, F. Mao, X.S. Li, F.Y. Kwong, Org. Lett. 13 (2011) 3258-3261;
(e) J. Park, E. Park, A. Kim, et al., Org. Lett. 13 (2011) 4390-4393;
(f) S. Sharma, E. Park, J. Park, I.S. Kim, Org. Lett. 14 (2012) 906-909;
(g) H.J. Li, P.H. Li, L. Wang, Org. Lett. 15 (2013) 620-623;
(h) F. Szabó, J. Daru, D. Simkó, et al., Adv. Synth. Catal. 355 (2013) 685-691;
(i) X.Y. Geng, C.Y. Wang, Org. Lett. 17 (2015) 2434-2437;
(j) Z.J. Cai, C. Yang, S.Y. Wang, S.J. Ji, J. Org. Chem. 80 (2015) 7928-7936;
(k)Y.F. Liang, X.Y.Wang, C.H.Tang, etal., Chem.Commun. (Camb.)52 (2016)1416-1419;
(l) X.P. Chen, X.L. Cui, Y.J. Wu, Org. Lett. 18 (2016) 2411-2414;
(m) P. Yang, Y.S. Bao, RSC Adv. 7 (2017) 53878-53886. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM19552400
(a) F.H. Xiao, Q. Shuai, F. Zhao, et al., Org. Lett. 13 (2011) 1614-1617;
(b) Y. Yuan, D.T. Chen, X.W. Wang, Adv. Synth. Catal. 353 (2011) 3373-3379;
(c) H. Tang, C. Qian, D.E. Lin, H.F. Jiang, W. Zeng, Adv. Synth. Catal. 356 (2014) 519-527;
(d) C.P. Perumgani, S.P. Parvathaneni, B. Kodicherla, S. Keesara, M.R. Mandapati, Inorg. Chim. Acta Rev. 455 (2017) 105-111.
(a) P. Fang, M.Z. Li, H.B. Ge, J. Am. Chem. Soc. 132 (2010) 11898-11899;
(b) M.Z. Li, H.B. Ge, Org. Lett. 12 (2010) 3464-3467;
(c) H. Wang, L.N. Guo, X.H. Duan, Org. Lett. 14 (2012) 4358-4361;
(d) J.Z. Yao, R.K. Feng, Z.H. Wu, Z.X. Liu, Y.H. Zhang, Adv. Synth. Catal. 355 (2013) 1517-1522;
(e) M. Kim, J. Park, S. Sharma, et al., Chem. Commun. 49 (2013) 925-927;
(f) Z.Y. Yang, X. Chen, J.D. Liu, et al., Chem. Commun. 49 (2013) 1560-1562;
(g) J. Park, M. Kim, S. Sharma, et al., Chem. Commun. 49 (2013) 1654-1656;
(h) C.D. Pan, H.M. Jin, X. Liu, Y.X. Cheng, C.J. Zhu, Chem. Commun. 49 (2013) 2933-2935;
(i) J.M. Miao, H.B. Ge, Org. Lett. 15 (2013) 2930-2933;
(j) Z.Y. Li, D.D. Li, G.W. Wang, J. Org. Chem. 78 (2013) 10414-10420;
(k) C. Zhou, P.H. Li, X.J. Zhu, L. Wang, Org. Lett. 17 (2015) 6198-6201;
(l) X.P. Chen, X.L. Cui, Y.J. Wu, Org. Lett. 18 (2016) 3722-3725;
(m) Y.N. Wu, L. Sun, Y.Y. Chen, et al., J. Org. Chem. 81 (2016) 1244-1250;
(n) P.Y. Lee, P. Liang, W.Y. Yu, Org. Lett. 19 (2017) 2082-2085;
(o) K. Jing, J.P. Yao, Z.Y. Li, et al., J. Org. Chem. 82 (2017) 12715-12725;
(p) Q.L. Li, Z.Y. Li, G.W. Wang, ACS Omega 3 (2018) 4187-4198. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM20698536
(a) W. Zhou, H.J. Li, L. Wang, Org. Lett. 14 (2012) 4594-4597;
(b) J.Y. Lu, H. Zhang, X.W. Chen, et al., Adv. Synth. Catal. 355 (2013) 529-536;
(c) P. Mamone, G. Danoun, L.J. Gooßen, Angew. Chem. Int. Ed. 52 (2013) 6704-6708;
(d) Q. Zhang, F. Yang, Y.J. Wu, Chem. Commun. 49 (2013) 6837-6839;
(e) S. Han, S. Sharma, J. Park, et al., J. Org. Chem. 79 (2014) 275-284.
(a) S. Guin, S.K. Rout, A. Banerjee, S. Nandi, B.K. Patel, Org. Lett.14 (2012) 5294-5297;
(b) Z.W. Yin, P.P. Sun, J. Org. Chem. 77 (2012) 11339-11344;
(c) F. Xiong, C. Qian, D.E. Lin, W. Zeng, X.X. Lu, Org. Lett. 15 (2013) 5444-5447;
(d) Y.N. Wu, P.Y. Choy, F. Mao, F.Y. Kwong, Chem. Commun. 49 (2013) 689-691;
(e) H.Y. Song, D. Chen, C. Pi, X.L. Cui, Y.J. Wu, J. Org. Chem. 79 (2014) 2955-2962.
(a) P.M. Liu, C.G. Frost, Org. Lett. 15 (2013) 5862-5865;
(b) A.B. Khemnar, B.M. Bhanage, Eur. J. Org. Chem. (2014) 6746-6752;
(c) B.W. Zhou, Y.Y. Hu, C.Y. Wang, Angew. Chem. Int. Ed. 54 (2015) 13659-13663.
(a) J. Wencel-Delord, F. Glorius, Nat. Chem. 5 (2013) 369-375;
(b) T. Cernak, K.D. Dykstra, S. Tyagarajan, P. Vachal, S.W. Krska, Chem. Soc. Rev. 45 (2016) 546-576;
(c) D.C. Blakemore, L. Castro, I. Churcher, et al., Nat. Chem. 10 (2018) 383-394;
(d) K.A. Margrey, W.L. Czaplyski, D.A. Nicewicz, E.J. Alexanian, J. Am. Chem. Soc. 140 (2018) 4213-4217;
(e) M.A. Brodney, R. Sharma, J.T. Lazzaro, G.S. Walker, R.S. Obach, Bioorg. Med. Chem. Lett. 28 (2018) 2068-2073;
(f) D.W. Yin, G. Liu, J. Org. Chem. 83 (2018) 3987-4001.
(a) C. Gao, H.C. Li, M.C. Liu, et al., RSC Rdv. 7 (2017) 46636-46643;
(b) L. Xu, C. Wang, Z.W. Gao, Y.M. Zhao, J. Am. Chem. Soc. 140 (2018) 5653-5658;
(c) X.B. Lu, Y.F. Shi, F.R. Zhong, Green Chem. 20 (2018) 113-117.
(a) H.X. Dai, A.F. Stepan, M.S. Plummer, Y.H. Zhang, J.Q. Yu, J. Am. Chem. Soc.133 (2011) 7222-7228;
(b) A.N. Lowell, M.D. DeMars, S.T. Slocum, et al., J. Am. Chem. Soc. 139 (2017) 7913-7920;
(c) F. Couly, C. Dubouilh-Benard, T. Besson, C. Fruit, Synthesis 49 (2017) 4615-4622.
(a) K. Beydoun, M. Zaarour, J.A.G. Williams, H. Doucet, V. Guerchais, Chem. Commun. 48 (2012) 1260-1262;
(b) Y. Kuninobu, S. Sueki, Synthesis 47 (2015) 3823-3845;
(c) Z. Zhang, P.H. Dixneuf, J.F. Soulé, Chem. Commun. 54 (2018) 7265-7280.
(a) A.A. Bekhit, T. Abdel-Aziem, Bioorg. Med. Chem. 12 (2004) 1935-1945;
(b) T.D. Paulis, K. Hemstapat, Y.L. Chen, et al., J. Med. Chem. 49 (2006) 3332-3344;
(c) S. Fustero, R. Román, J.F. Sanz-Cervera, et al., J. Org. Chem. 73 (2008) 8545-8552;
(d) C.E. Mowbray, C. Burt, R. Corbau, et al., Bioorg. Med. Chem. Lett. 19 (2009) 5857-5860;
(e) G.P. Lahm, D. Cordova, J.D. Barry, Bioorg. Med. Chem. 17 (2009) 4127-4133;
(f) A.K. Ghosh, M. Brindisi, Y.C. Yen, et al., Bioorg. Med. Chem. Lett. 25 (2015) 668-672;
(g) G. Alvarez, J. Varela, E. Cruces, et al., Antimicrob. Agents Chemother. 59 (2015) 1398-1404;
(h) C. Dockendorff, P.W. Faloon, M. Yu, et al., ACS Med. Chem. Lett. 6 (2015) 375-380.
(a) X.M. Fan, Y. Guo, Y.D. Li, et al., Asian J. Org. Chem. 5 (2016) 499-505;
(b) K.K. Yu, Y. Guo, Y.H. Hu, et al., Asian J. Org. Chem. 5 (2016) 1219-1224.
R.Y. Zhu, L.Y. Liu, H.S. Park, et al., J. Am. Chem. Soc. 139 (2017) 16080-16083. doi: 10.1021/jacs.7b09761
(a) J.J. Warren, J.M. Mayer, J. Am. Chem. Soc. 132 (2010) 7784-7793;
(b) W. Ali, A. Behera, S. Guin, B.K. Patel, J. Org. Chem. 80 (2015) 5625-5632.
Z. Ren, G.B. Dong, Organometallics 35 (2016) 1057-1059. doi: 10.1021/acs.organomet.6b00185
(a) Q. Liu, R. Jackstell, M. Beller, Angew. Chem. Int. Ed. 52 (2013) 13871-13873;
(b) N.R. Deprez, M.S. Sanford, J. Am. Chem. Soc. 131 (2009) 11234-11241.

Figure 1 Directing effect of the substituted pyrazoles. Reaction conditions: 1 (0.3 mmol), 2a (63.7mg, 0.6 mmol), Pd(TFA)2 (10.0 mg, 10 mol%), TBHP (67.6mg, 2.5 equiv.), DCE (2.0 mL), N2 atmosphere, 90 ℃, 12h. Isolated yields of 3. DCE =1, 2- dichloroethane; TBHP = tert-butyl hydroperoxide.

Figure 3 Substrate scope of functionalized pyrazoles. Reaction conditions: 1 (0.3 mmol), 2a (63.7 mg, 0.6 mmol), Pd(TFA)2 (10.0 mg, 10 mol%), TBHP (67.6 mg, 2.5 equiv.), DCE (2.0 mL), 100 ℃, 12 h in N2 atmosphere. Isolated yields.

Figure 4 Substrate scope of arylaldehydes. Reaction conditions: 1g (100.9 mg, 0.3 mmol), 2 (0.6 mmol), Pd(TFA)2 (10.0 mg, 10 mol%), TBHP (67.6 mg, 2.5 equiv.), DCE (2.0 mL), 100 ℃, 12 h in N2 atmosphere. Isolated yields.

Scheme 4 Preparation and functionlization of the dual-core dimeric cyclopalladated intermediate I.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



