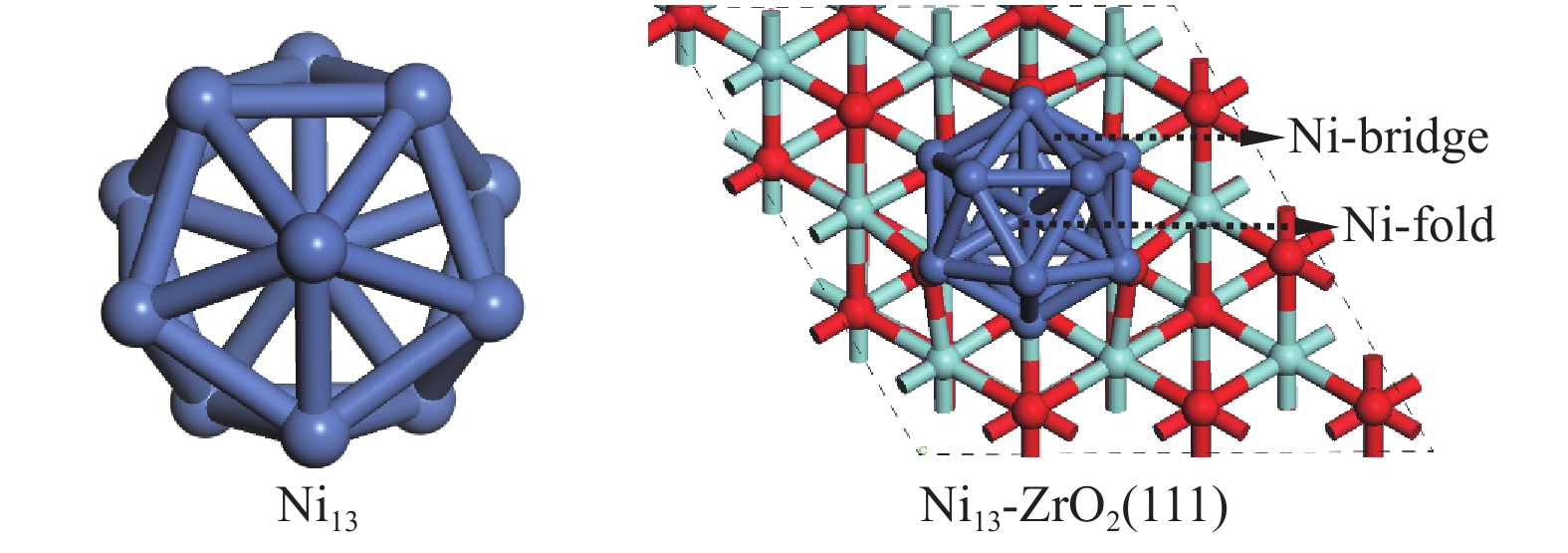
Figure 1.
Top view correspond to Ni13 and Ni13-ZrO2(111) morphologies, respectively
Unraveling the role of Ni13 catalyst supported on ZrO2 for CH4 dehydrogenation: The d-band electron reservoir
Cui-mei ZHI , Rui-hong YANG , Chang-yu ZHOU , Gui-ru WANG , Jia-ying DING , Wen YANG

With global warming and the depletion of crude oil reserves, the development of alternative resources attracted more attention over the past decades[1]. Methane has been regarded as an important clean energy and it can be converted to various commodity chemicals[2]. The activation of methane to form CHx through C−H bond breaking is the key [3-11]. Hence, many efforts have been carried out to develop and design efficient catalyst for the activation of CH4.
Transition metals such as Rh[3], La[4] and Ni[5-11] possess a remarkable catalytic activity for methane dissociation. Typically, supported Ni-based catalysts are of much interest, because of their high activity and low costs. For methane decomposition, the activity and dispersion of Ni are crucial. In other words, the more exposed sites in Ni-based catalyst, the higher reactivity.
Firstly, the activation of C−H bond in methane needs additional electrons. Due to the abundant d orbitals, metal oxide support exhibits better electron donation ability that enhances the reactivity of Ni. Secondly, the support with higher specific surface area is extremely benefit to disperse and anchor the fine Ni particles. This has been confirmed by previous reported literatures[12-16]. Therefore, loading Ni onto a proper support can provide additional electronic property and improve the dispersion of Ni, thereby promoting the dissociation of CH4.
In particular, ZrO2 is known to have a superior electronic donor ability to enhance the Ni catalytic activity[12], although ZrO2 itself has no activity for CH4 dissociation[17,18]. Wang et al.[12] concluded that the Ni/ZrO2 catalyst can stabilize the original Ni state due to its small size; the strong electronic donor ability of ZrO2 was benefit to the formation of Ni active particles in carbon dioxide reforming of methane. Besides, the introduction of Zr ensured an excellent dispersion of Ni/Zr alloy and promoted the formation of active center, both of which facilitated the activation of the C–H bond in CH4[19], and thus, decreasing the Ea value in dry reforming of methane. Similarly, Han et al.[13] pointed out that in addition to facilitating the Ni dispersion, support can also alter the electronic property of the catalysts that influences the adsorption characteristics, the activation energy and even the reaction pathways.
In fact, the influence of strong interaction between Ni and ZrO2 on the activity and dispersion of Ni has been confirmed by the recent reports[19-23]. Thanks to the strong interaction between the support ZrO2 and Ni catalyst, the dispersion of Ni active components is considerably enhanced[13,17,19-23]. Besides, the interaction between nickel and support might maintain the initial state of Ni[12]. Meanwhile, the moderate interaction, generated from the partial electron transfer between Ni and Zr, might promote the adsorptions of CHx[12]. Li et al.[24] believed that La-modified Ni/Al2O3 is responsible for improving the stability and reactivity of Ni catalyst via modulating the interaction between Ni and Al2O3.
Since the support has significant influence on the activity of Ni-based catalyst in CH4 activation, it is necessary to investigate the interaction of nickel and support and the electron transfer between them. It is shown that the catalytic activity of small cluster Ni13 supported on ZrO2(111) surface differs from that of large Ni(111) in the CO2 hydrogenation reaction due to their different surface atomic arrangement and electronic characteristics[25]. Ni13 exhibits higher catalytic activity and selectivity, as it has larger number of exposed surface atoms and more active sites with different coordination numbers[26]. Ni13 has stable icosahedron configurations that contains 42 Ni–Ni bonds[27,28]. This makes Ni13 can well maintain the structure during catalysis process[29]. Ni atom in the center of Ni13 shows the highest coordination number, while each Vertex Ni atom of Ni13, possessing the significantly coordinatively unsaturated site, is the predominant active center for Ni catalyst. Yilmazer’s[30] stated that ethylene adsorption energy gradually increases with the decrease of Ni coordination number from 9 to 6 on Ni(111), Ni(100), Ni(110) and Ni13. Therefore, the icosahedral Ni13 with Ih symmetry shows higher the structural stability and reactivity[31,32] among different Nin[33].
Hence, the reaction mechanism for CH4 dehydrogenation on pure Ni13 and Ni13 supported ZrO2(111) catalysts was investigated by using DFT calculations. The effect of support on the adsorption and activation of CHx (x=0−3) was analyzed. The results suggested that the interaction between Ni and ZrO2 can improve the electron delocalization of Ni and make the adsorbed CHx (x=0−3) species become more electron-rich in reaction.
DFT calculations were performed using the Vienna Ab Initio Simulation Package (VASP), where the exchange-correlation energy functional is described with Perdew-Burke-Ernzerhof (PBE) of the generalized gradient approximation (GGA)[34]. The configurations of Ni13 and Ni13-ZrO2(111) are shown in Figure 1. Ni13 is obtained on the basis of the magic number cluster structure[30], the average Ni–Ni distance in Vertex-Vertex (2.444 Å) and in Center-Vertex (2.325 Å) are in line with the experimental results (2.389 and 2.274 Å, respectively)[35] and theoretical calculation[26]. The p (3×3) slabs ZrO2(111) has nine layers. A vacuum of 15 Å is added, and a lattice parameter of 5.13 Å is used[36-38]. The top five layers are fully relaxed, while the bottom four layers are fixed at their bulk positions. The energy convergence for electronic relaxation is set to 10−5 eV. All atoms are relaxed until the atomic force is less than 0.02 eV/Å with a kinetic energy cutoff of 400 eV[39]. The Monkhorst-Pack 2×2×1 k-point mesh is used to Brillouin zones of sample surface[40]. The transition state is searched by using the nudged elastic band method , as described by our previous work[41].
For the adsorptions and reactions on Ni13 and Ni13-ZrO2(111), the adsorption energies (Eads), the activation energy (Ea) and reaction energy (ΔE) with the zero-point-energy (ZPE) are defined according to equations (1), (2) and (3)[41,42], where Ecluster, Eadsorbate and Eadsorbate/cluster are the total energies of the bare cluster, the isolated adsorbate, and the optimized structures of the adsorption configuration, respectively. ΔZPEads, ΔZPEbarrier and ΔZPEreaction refer to ZPE corrections for Eads, Ea and ΔE, respectively, which are determined according to equations (4), (5) and (6)[41,42]; h is Planck's constant, υi refers to the vibrational frequencies. The d-band center (
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
|
|
(6) |
|
|
(7) |
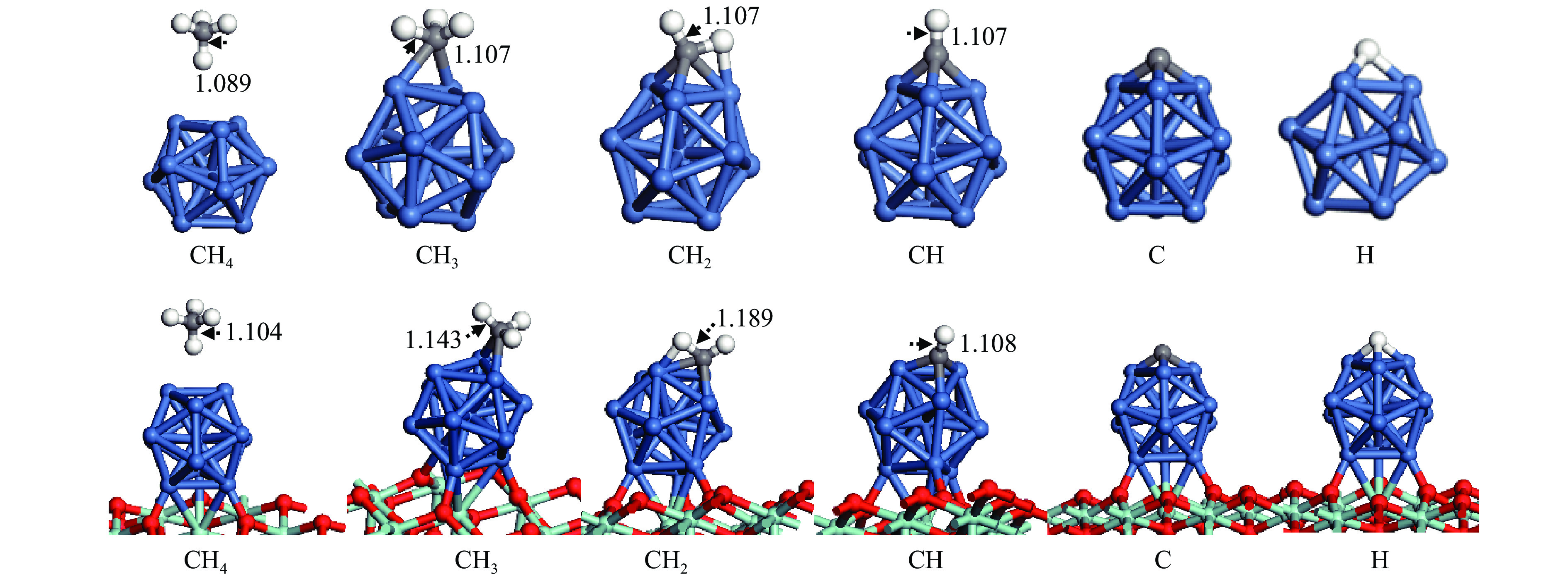
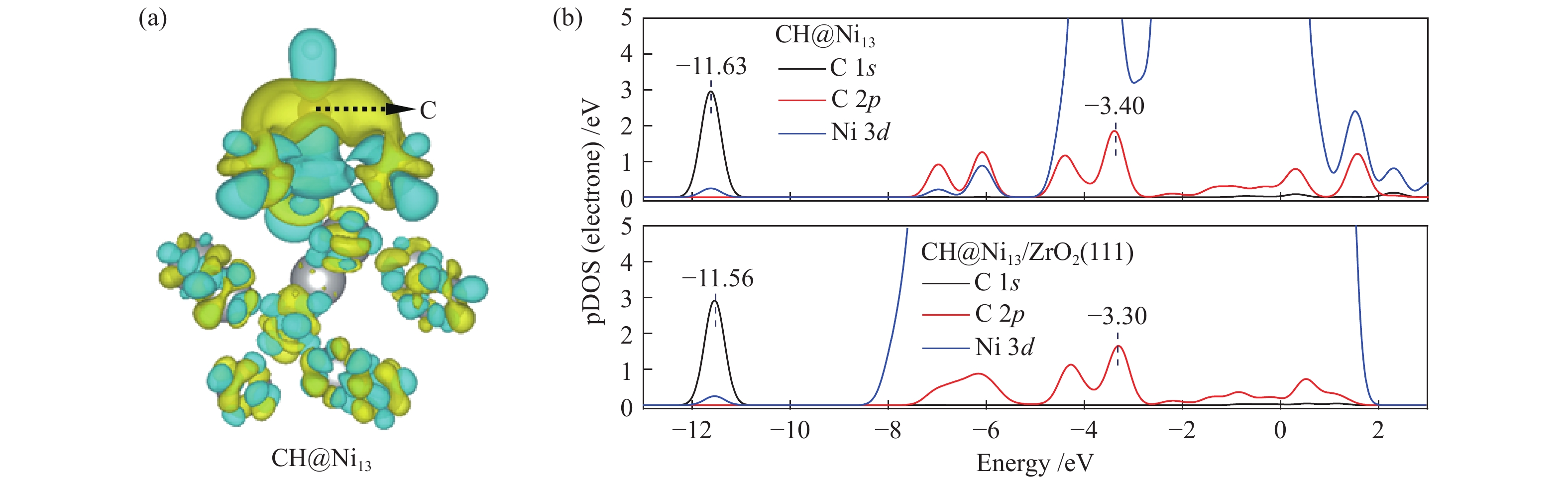
The optimization structure in DFT calculation and corresponding energy of all adsorbed species involving in CH4 dissociation on Ni13 and Ni13-ZrO2(111) are presented in Figure 2 and Table 1. For comparison, previous calculated results on Ni(100) and Ni4 are also given in Table 1. The partial density of states (pDOS) and the differential charge density for CH adsorbed on Ni13 and Ni13-ZrO2(111) are shown in Figure 3.
 下载:
导出CSV
下载:
导出CSV
| Species | Ni13 | Previous results Ni4[6], Ni(100)[7] | Ni13-ZrO2(111) | |||
| site | Eads/eV | Eads/eV | site | Eads/eV | ||
| CH4 | Ni-top | 0.04 | −0.41[6] | Ni-top | −0.02 | |
| CH3 | Ni-bridge | −2.22 | −2.37[6] | Ni-bridge | −2.46 | |
| CH2 | Ni-fold | −4.36 | −4.85[6], −3.76[7] | Ni-fold | −4.76 | |
| CH | Ni-fold | −6.40 | −6.71[6], −6.43[7] | Ni-fold | −7.00 | |
| C | Ni-fold | −7.21 | −8.94 [6], −7.27[7] | Ni-fold | −7.96 | |
| H | Ni-fold | −2.78 | −2.41[6], −2.36[7] | Ni-fold | −3.02 | |
| DFT methods: Gaussian 09W code, GGA-PBE[6]; ADF-BAND code, GGA-RPBE[7] | ||||||
As shown in Figure 2 and Table 1, CH4 is weakly adsorbed at Ni-top of Ni13 and Ni13-ZrO2(111) with similar adsorption energies of 0.04 and −0.02 eV, respectively. The physisorption nature of CH4 arises from the van der Waals interaction between CH4 and catalyst. The calculated CH4 adsorption energy is in agreement with the results of An et al. (−0.01 eV) [43], despite that Roy and co-workers gave a more negative value (−0.41 eV)[6].
The configurations for CH3, CH2, CH, C and H adsorption on Ni13-ZrO2(111) are quite similar to those on Ni13. CH3 adsorbed at the Ni-bridge site of Ni13-ZrO2(111) is more stable than at Ni13; notably, the calculated CH3 adsorption energy (−2.46 eV) in this work is similar to previous results (−2.37 eV), proving the credibility of current calculations[6]. CH2, CH, C and H prefer to adsorb at Ni-fold sites (Table 1), and they give higher adsorption energy on Ni13-ZrO2(111) than on Ni13. Moreover, the calculated adsorption energies of CH2 and CH (−4.76 and −7.00 eV) on Ni13-ZrO2(111) are also consistent with the reported values (−4.85 and −6.71 eV) on Ni4 by Roy et al. (Table 1)[6]. But the C is strongly adsorbed in four-fold hollow site with adsorption energy of −8.94 eV [6] than in three-fold hollow site on Ni13-ZrO2(111) (−7.96 eV), thus signifying that C atom tends to satisfy its valence. It should be noticed that the adsorption energies of CH2 (−3.76 eV), CH (−6.43 eV), C (−7.27 eV) and H (−2.36 eV) on Ni(100) obtained by Li et al.[7] are different to our results (−4.76, −7.00, −7.96 and −3.02 eV) on Ni13-ZrO2(111). This may be due to the use of different functional.[7]
Despite the similarity in adsorption configuration and site, the adsorption strength of all these species are stronger on Ni13-ZrO2(111) than on Ni13, possibly due to the existence of ZrO2 support that modifies the electronic environment of Ni13. For example, for CH adsorption, as shown in Figure 3(a), the electron accepting behavior of C and charge loss behavior of Ni occur in the yellow areas and in the scattered blue areas, respectively. An accumulation of electron density is observed on the C atom, whereas for Ni atoms bonding to C atom, the electron density is decreased. Thus, the electron transfer between Ni→C turns the chemisorbed state of free CHx[44].
As shown in Figure 3(b), with the addition of ZrO2 support, C 1s and C 2p orbitals are shifted to higher value, while the Ni 3d orbital gives an opposite result. This leads to a larger overlap between C 2p and Ni 3d as well as a stronger interaction between Ni and C atoms. Therefore, a strong adsorption of CH on Ni13-ZrO2(111) is observed. Similar result was also observed by Li et al.[7]: the adsorption strength of C on various Ni surfaces dependents on the mixing between C 2p and Ni 3d orbitals. Furthermore, the more downshift the Ni 3d orbital is, the more stable adsorption of CHx is[34].
In addition, as shown in Figure 2, C−H bond distance for CH4 (1.104 Å), CH3 (1.143 Å), CH2 (1.189 Å) and CH (1.108 Å) are significantly enlarged on Ni13-ZrO2(111), compared to that on Ni13 (1.089, 1.107, 1.107 and 1.107 Å, respectively). This means that the cleavage of C−H bonds in CHx is easier on Ni13-ZrO2(111).
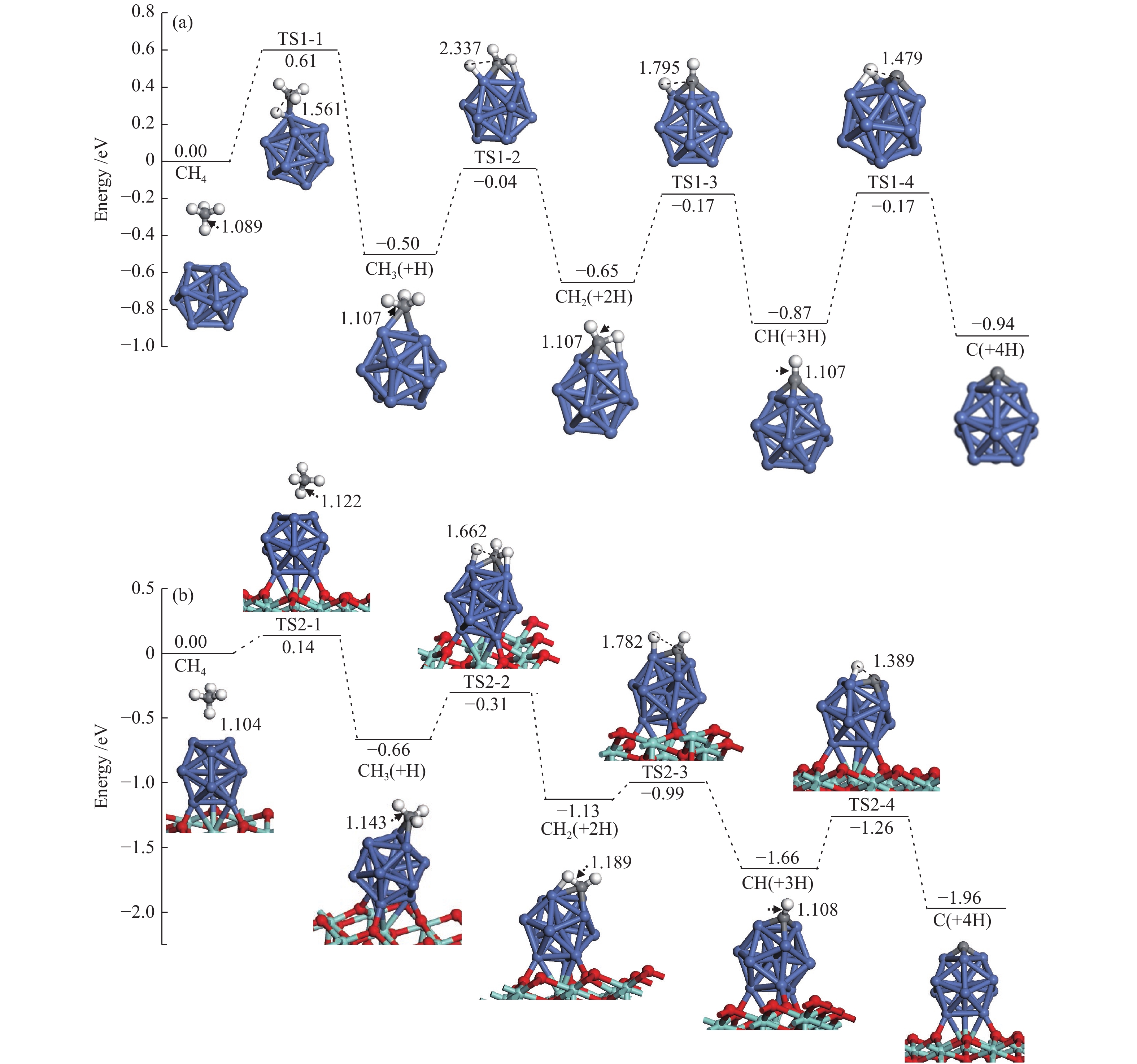
The corresponding energies of various elementary steps for CH4 dissociation are listed in Table 2. Previously reported results on Ni(100) and Ni4 are also given in Table 2 for comparison. The energy profile and the main configurations for the initial state (ISs), transition state (TSs) and final states (FSs) on Ni13 and Ni13-ZrO2(111) are displayed in Figure 4(a) and 4(b), respectively. The values for C2 formation and C elimination are calculated in Figure 5.
下载:
导出CSV
| Reaction | Our results | Previous results | ||||||||
| Ea/eV | ΔE/eV | v/cm−1 | dC–H ISs/Å | dC–H TSs/Å | H displacements/Å | Ea/eV | ΔE/eV | |||
| Ni13 | Ni4[6] | Ni(100)[7] | ||||||||
| CH4→CH3+H | R1-1 | 0.61 | −0.50 | 932 i | 1.089 | 1.561 | 0.472 | 1.23[7] | ||
| CH3→CH2+H | R1-2 | 0.46 | −0.15 | 416 i | 1.107 | 2.337 | 1.230 | 0.84[6], 0.62[7] | −1.41[6] | |
| CH2→CH+H | R1-3 | 0.48 | −0.22 | 625 i | 1.107 | 1.795 | 0.688 | 0.33[6], 0.22[7] | −4.40[6] | |
| CH→C+H | R1-4 | 0.70 | −0.07 | 747 i | 1.107 | 1.479 | 0.372 | 1.37[6], 0.64[7] | −5.96[6] | |
| C+C→C2 | R1-5 | 3.80 | 0.62 | 524 i | ||||||
| Ni13-ZrO2(111) | ||||||||||
| CH4→CH3+H | R2-1 | 0.14 | −0.66 | 59 i | 1.104 | 1.122 | 0.018 | |||
| CH3→CH2+H | R2-2 | 0.35 | −0.47 | 760 i | 1.143 | 1.662 | 0.519 | |||
| CH2→CH+H | R2-3 | 0.14 | −0.53 | 488 i | 1.189 | 1.782 | 0.593 | |||
| CH→C+H | R2-4 | 0.40 | −0.30 | 657 i | 1.108 | 1.389 | 0.281 | |||
| C+C→C2 | R2-5 | 1.90 | 1.87 | 170 i | ||||||
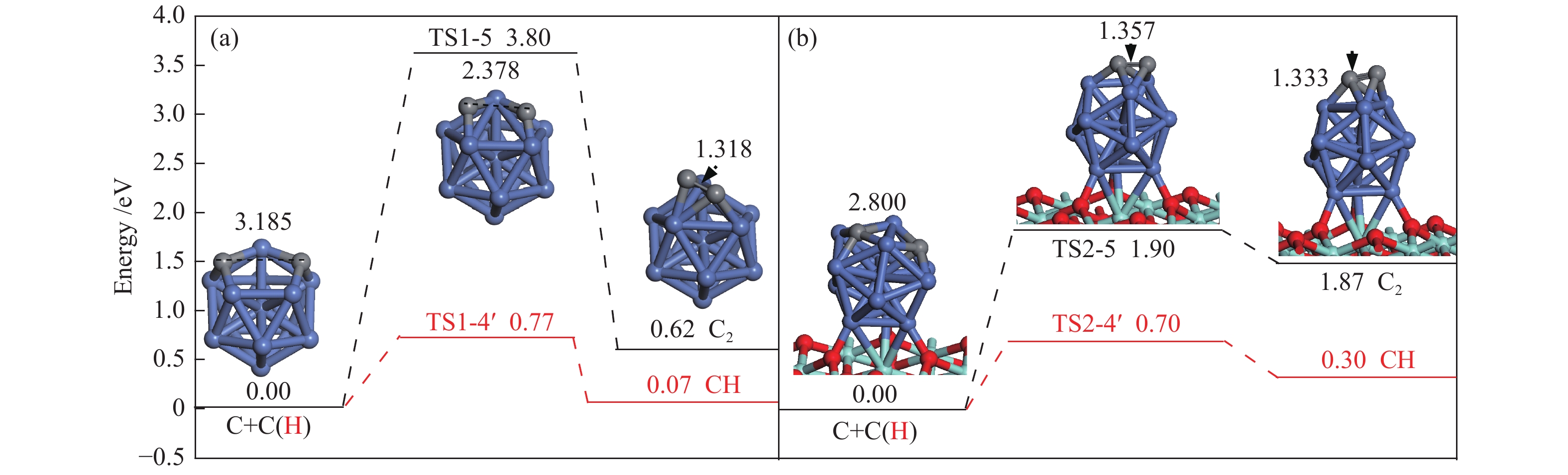
Bond lengths are in Å see Figure 2 for color coding
We mainly focus on the Ea , ΔE and the C−H bonds length of ISs and TSs. The activation energies for the dehydrogenation of CHx (x=4−1) on Ni13-ZrO2(111) are lower than on Ni13, indicating that the dehydrogenation of CHx is kinetically more favorable on the former than on the latter.
As shown in Table 2, the calculated activation energy ( Ea , 0.61 eV) on Ni13 is lower than the reported result (1.23 eV) by Li et al.[7] on Ni(100), whereas that for CH3 (0.46 eV) and CH2 (0.48 eV) dehydrogenations are similar to the obtained values by Li et al. (0.62 and 0.22 eV)[7] and Roy et al. (0.84 and 0.33 eV)[6]. For CH dehydrogenation, the activation energy on Ni13 (0.70 eV) and on Ni(100) (0.64 eV) are similar, but both of them are much lower than on Ni4 cluster (1.37 eV). It should be noticed that the dissociations of CHx (x=4−1) on Ni13-ZrO2(111) are all exothermic, with more negative ΔE than Ni13.
For CH4→CH3+H on Ni13-ZrO2(111), it shows a low Ea of 0.14 eV, with exothermic by −0.66 eV; the C–H bond is increased from 1.104 Å in CH4 to 1.122 Å in TS2-1 . After a rearrangement between H and CH3 fragments, the structure of TS2-1 is obtained. Similar procedure is used for TS1-1 , despite that the Ea is increased to 0.61 eV, with a weak exothermic by −0.50 eV on Ni13. The C–H distance of TS1-1 is enlarged from 1.089 Å to 1.561 Å. Since the dissociation of C−H bond is more exothermic on Ni13-ZrO2(111) (TS2-1), it leads to the reduction of Ea[45]. Accordingly, the ZrO2 support facilitates CH4→CH3+H, due to the strong interaction between Ni and ZrO2, being line with previous investigation[17].
Similar to CH4→CH3+H, stepwise dehydrogenations of CH3 to CH2, CH and C show low Ea values of 0.35, 0.14 and 0.40 eV for TS2-2 , TS2-3 and TS2-4 , with exothermic by −0.47, −0.53 and −0.30 eV, respectively, on Ni13-ZrO2(111). The displacements between the detaching H and the remaining fragment are 0.519, 0.593 and 0.281 Å, respectively, as shown in Table 2. However, without ZrO2, the activation energy of TS1-2 , TS1-3 and TS1-4 is elevated to 0.46, 0.48 and 0.70 eV, along with the reduction of exothermic energy to −0.15, −0.22 and −0.07 eV, respectively. Meanwhile, the displacements between H and remaining fragments are also increased to 1.230, 0.688 and 0.372 Å, respectively (Table 2). Thus, the ZrO2 support has a significant influence on CH4 dissociation. This observation is consistent with the previous study[12] where the ZrO2 support can modulate the electronic environment of Ni and promote the dissociation of C−H bond.
Based on Ea and ΔE of CH4 stepwise dehydrogenations to C, the overall activation energies of CH4 dissociation on Ni13 and Ni13-ZrO2(111) are calculated to be 0.61 and 0.14 eV, respectively, as plotted in Figure 4(a) and 4(b). This gives additional evidence that ZrO2 support effectively promotes CH4 dissociation. Similar situation is also observed in previous reports[12,19]. As stated by Han et al.[13], this is because the support can tune the adsorption behaviors, decrease the activation barrier and even adjust pathways of dry reforming of methane by modifying the Ni catalyst.
Once C is formed, C nucleation may lead to coke deposition. Interestingly, as shown in Figure 5, C+H→CH, as the reverse steps of CH scission, is preferred to C+C→C2 on Ni13 and Ni13-ZrO2(111), suggesting that carbon deposition is unlikely to occur on both catalyst surfaces. Furthermore, the activation energy ( Ea ) of 1.90 eV for C+C→C2 on Ni13-ZrO2(111) is similar to that of C2 dimmer formation (1.89 eV)[46]. Therefore, the undesired coke deposition may be observed only when the residual C can not be timely removed from the catalyst surface[47]. Thus, both the highly dispersed Ni13 and the superior electronic donor ability of ZrO2 support suppress the coke deposition. This is because C nucleation is inferior to C elimination.
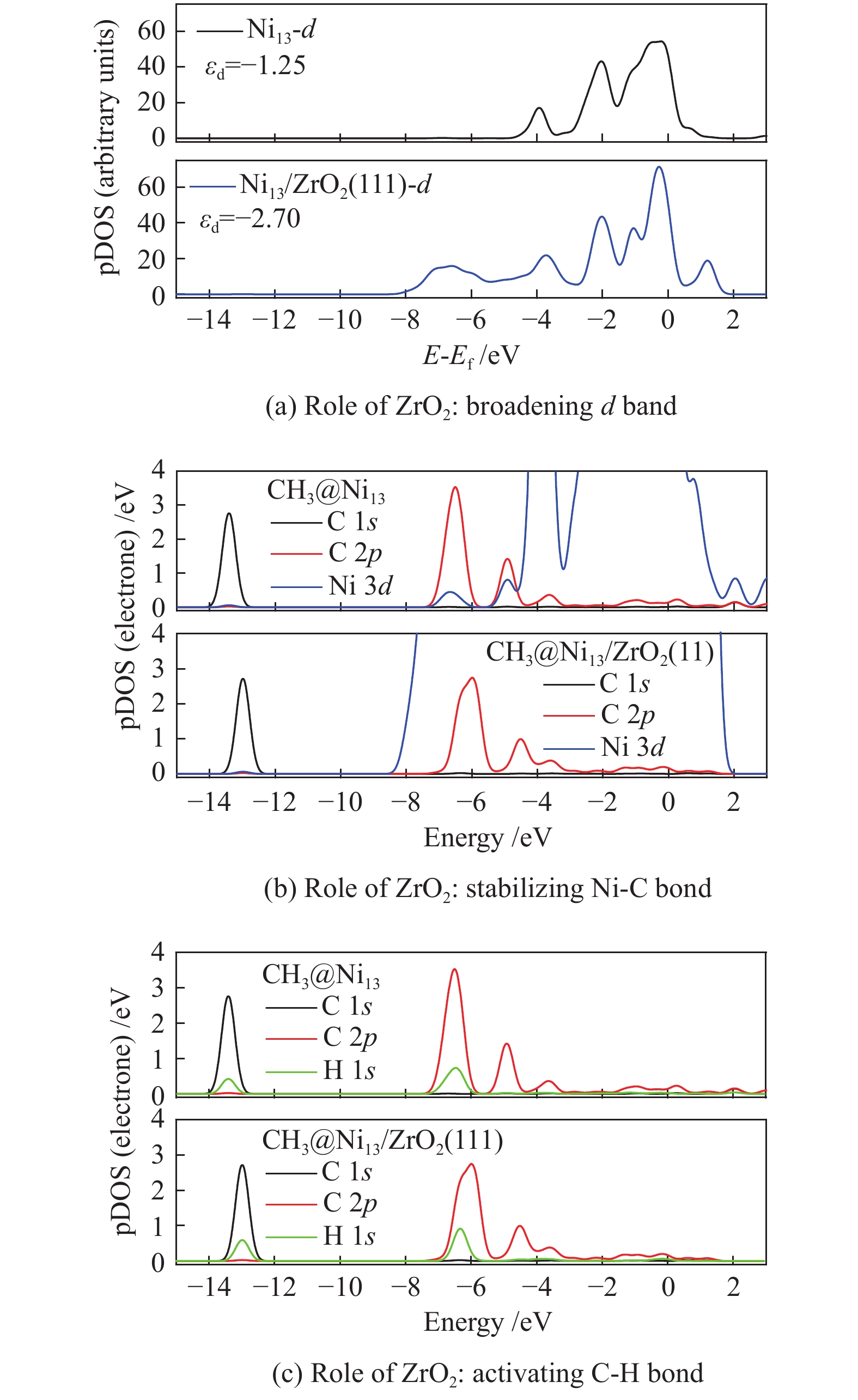
The most important steps in CH4 dissociation are adsorption and dissociation, which leads to the formation of Ni–C bond and the dissociation of C−H bond, respectively. To further explore the effect of ZrO2 support on CH4 decomposition, the pDOS of CH3 on Ni13 and Ni13-ZrO2(111), as an example, is calculated in Figure 6. Accordingly, the role of ZrO2 support in CH4 dissociation is obtained.
The result of electronic analysis indicates that the presence of ZrO2 support broadens the d-band electrons at C of CH3 bonding on Ni (Figure 6(a)). This causes a larger overlap between C 2p and Ni 3d, i.e. and a stronger interaction between Ni and C (Figure 6(b)), thereby leading to more stable adsorptions of CHx (x=3−1) and higher exothermic energy for CH4 dehydrogenation. As stated by Boukelkoul et al.[48], CH4 dehydrogenation on W-Cu(100) is exothermic because W-doping stabilizes better for all species. Ou et al.[44] also proved that the adsorption state becomes highly stable, when more electrons are transferred from Ni to C of CHx .
Besides, the charge distribution on C and H is influenced due to the enhancement of electron transfer Ni→C on Ni13-ZrO2(111), whereas that of H→C is reduced. This causes a weaker C 2p −H 1s hybridization than the situation on Ni13, as seen in Figure 6(c), thereby decreasing the Ea for C−H bond breaking. Similar results[44] are observed on Co/Ni, where the low activation energy for C−H bond dissociation in CH4 dehydrogenation resulted from the abundant d-band electrons on Co/Ni catalyst.
As a result, the overall activation energy for CH4 dissociation drastically decreases with the addition of ZrO2 support. This is because ZrO2 support can act as the d-band electron reservoir at Ni13 that promotes the activation of C−H bond in CH4 dehydrogenation reactions. As stated by Ou et al.[44], introduction of Co into Ni leaded to the downshift of d-band center of Co/Ni that increased the Eads of the CHx , but decreased the Ea of C−H bond breakage. These results are further supported by the work Li et al.[8]; the strong C−Ni chemical bonding can electronically adjust the ability of Ni atoms in adsorbing and dissociating CHx (x=4−1) species.
DFT calculations are used to investigate the dehydrogenation mechanism of CH4 on Ni13 and Ni13-ZrO2(111). The results show that ZrO2 support has a significant influence on promoting CH4 dissociation.
The Eads, Ea, ΔE and the displacements of the detaching H are obtained. Although the configuration and adsorption site are similar, the adsorptions of all species involving in CH4 dissociation are stronger on Ni13-ZrO2(111) than on Ni13. This is because the ZrO2 support remarkably modifies the electronic environment of Ni13. Besides, introduction of the ZrO2 support can obviously decrease Ea and simultaneously increase ΔE for all the elementary steps in CH4 dissociation. In particular, the Ea for conversion of CH4 to CH3 is reduced from 0.61 eV on Ni13 to 0.14 eV on Ni13-ZrO2(111). This results in the decrease of overall activation energy for CH4 dissociation on Ni13-ZrO2(111) than on Ni13. Thus, the ZrO2 modified Ni13 can improve the activity towards CH4 dissociation, compared with the pure Ni13.
Moreover, the C−H bonds distance in the ISs of CHx (x=4−1) dissociation is significantly enlarged when the ZrO2 support is added. This implies that the cleavage of C−H bonds of CHx (x=4−1) becomes easier. Based on TSs , the stepwise detachment of H from CH4 to CH3, CH2, CH and C needs to shift smaller distances to reach the final state. This also benefits to decrease the activation energy ( Ea ) of C−H bond dissociation.
In viewpoint of the electronic environment, the ZrO2 support can broaden d-band electrons at C atom bonding to Ni that enhances the electron transfer Ni→C, but reduces that of H→C. This causes a stronger interaction between Ni and C, along with the weakness of C−H polar bonds, thereby leading to a strong adsorption of CHx (x=3−1) and a low Ea for C−H bond dissociation. Hence, the presence of ZrO2 support can stabilize the Ni–C bond and activate the C−H bond. The mechanistic investigation of CH4 dehydrogenation over Ni13 and Ni13-ZrO2(111) may provide an approach to reveal the role of ZrO2 support and design new Ni-based catalysts for CH4 dissociation.
STOLAROFF J K, BHATTACHARYYA S, SMITH C A, BOURCIER W L, CAMERON-SMITH P J, AINES R D. Review of methane mitigation technologies with application to rapid release of methane from the arctic[J]. Environ Sci Technol,2012,46(12):6455−6469. doi: 10.1021/es204686w
CABALLERO A, PÉREZ P J. Methane as raw material in synthetic chemistry: The final frontier[J]. Chem Soc Rev,2013,42(23):8809−8820. doi: 10.1039/c3cs60120j
GROOTEL P W V, SANTEN R A V, HENSEN E J M. Methane dissociation on high and low indices Rh surfaces[J]. J Phys Chem C,2011,115(26):13027−13034. doi: 10.1021/jp2033774
LI B, METIU H. Dissociation of methane on La2O3 surfaces doped with Cu, Mg, or Zn[J]. J Phys Chem C,2011,115(37):18239−18246. doi: 10.1021/jp2049603
ZHANG T Y, HOLIHARIMANANA D, YANG X F, GE Q F. DFT study of methane activation and coupling on the (0001) and (112̅0) surfaces of α-WC[J]. J Phys Chem C,2020,124(49):26722−26729. doi: 10.1021/acs.jpcc.0c06928
ROY G, CHATTOPADHYAY A P. Dissociation of methane on Ni4 cluster-A DFT study[J]. Comput Theor Chem,2017,1106:7−14. doi: 10.1016/j.comptc.2017.02.030
LI J D, CROISET E, RICARDEZ-SANDOVAL L. Methane dissociation on Ni(100), Ni(111), and Ni(553): A comparative density functional theory study[J]. J Mol Catal A-Chem,2012,365:103−114. doi: 10.1016/j.molcata.2012.08.016
LI J D, CROISET E, RICARDEZ-SANDOVAL L. Effect of carbon on the Ni catalyzed methane cracking reaction: A DFT study[J]. Appl Surf Sci,2014,311:435−442. doi: 10.1016/j.apsusc.2014.05.081
VASILIADES M A, DJINOVIĆ P, PINTAR A, KOVAČ J, EFSTATHIOU A M. The effect of CeO2-ZrO2 structural differences on the origin and reactivity of carbon formed during methane dry reforming over NiCo/CeO2-ZrO2 catalysts studied by transient techniques[J]. Catal Sci Technol,2017,7:5422−5434. doi: 10.1039/C7CY01009E
ZHANG S S, YING M, YU J, ZHAN W C, WANG L, GUO Y, GUO Y L. NixAl1O2-δ mesoporous catalysts for dry reforming of methane: The special role of NiAl2O4 spinel phase and its reaction mechanism[J]. Appl Catal B: Environ,2021,291:120074. doi: 10.1016/j.apcatb.2021.120074
ZHANG L, WANG X G, SHANG X F, TAN M W, DING W Z, LU X G. Carbon dioxide reforming of methane over mesoporous nickel aluminate/γ-alumina composites[J]. J Energy Chem,2017,26(1):93−100. doi: 10.1016/j.jechem.2016.08.001
WANG Y, YAO L, WANG Y N, WANG S H, ZHAO Q, MAO D H, HU C W. Low-temperature catalytic CO2 dry reforming of methane on Ni-Si/ZrO2 catalyst[J]. ACS Catal,2018,8(7):6495−6506. doi: 10.1021/acscatal.8b00584
HAN J W, PARK J S, CHOI M S, LEE H. Uncoupling the size and support effects of Ni catalysts for dry reforming of methane[J]. Appl Catal B: Environ,2017,203:625−632. doi: 10.1016/j.apcatb.2016.10.069
BAUDOUIN D, RODEMERCK U, KRUMEICH F, MALLMANN A D, SZETO K C, MÉNARD H, VEYRE L, CANDY J P, WEBB P B, THIEULEUX C, COPÉRET C. Particle size effect in the low temperature reforming of methane by carbon dioxide on silica-supported Ni nanoparticles[J]. J Catal,2013,297:27−34. doi: 10.1016/j.jcat.2012.09.011
CUI Y H, XU H Y, GE Q J, WANG Y Z, HOU S F, LI W Z. Structure sensitive dissociation of CH4 on Ni/α-Al2O3: Ni nano-scale particles linearly compensate the Ea and ln A for the CH4 pulse kinetics[J]. J Mol Catal A-Chem,2006,249(1/2):53−59. doi: 10.1016/j.molcata.2006.01.009
YAN X L, HU T, LIU P, LI S, ZHAO B R, ZHANG Q, JIAO W Y, CHEN S, WANG P F, LU JJ, FAN L M, DENG X N, PAN Y X. Highly efficient and stable Ni/CeO2-SiO2 catalyst for dry reforming of methane: Effect of interfacial structure of Ni/CeO2 on SiO2[J]. Appl Catal B Environ,2019,246:221−231. doi: 10.1016/j.apcatb.2019.01.070
KAMBOLIS A, MATRALIS H, TROVARELLI A, PAPADOPOULOU C. Ni/CeO2-ZrO2 catalysts for the dry reforming of methane[J]. Appl Catal A: Gen,2010,377(1/2):16−26. doi: 10.1016/j.apcata.2010.01.013
DJINOVIĆ P, ČRNIVEC I G O, ERJAVEC B, PINTAR A. Influence of active metal loading and oxygen mobility on coke-free dry reforming of Ni-Co bimetallic catalysts[J]. Appl Catal B: Environ,2012,125:259−270. doi: 10.1016/j.apcatb.2012.05.049
YAO L, SHI J, XU H L, SHEN W, HU C W. Low-temperature CO2 reforming of methane on Zr-promoted Ni/SiO2 catalyst[J]. Fuel Process Technol,2016,144:1−7. doi: 10.1016/j.fuproc.2015.12.009
ZHANG M, ZHANG J F, ZHOU Z L, CHEN S Y, ZHANG T, SONG F E, ZHANG Q D, TSUBAKI N, TAN Y S, HAN Y Z. Effects of the surface adsorbed oxygen species tuned by rare-earth metal doping on dry reforming of methane over Ni/ZrO2 catalyst[J]. Appl Catal B: Environ,2020,264:118522. doi: 10.1016/j.apcatb.2019.118522
DĘBEK R, GALVEZ M E, LAUNAY F, MOTAK M, GRZYBEK T, COSTA P D. Low temperature dry methane reforming over Ce, Zr and CeZr promoted Ni-Mg-Al hydrotalcite-derived catalysts[J]. Int J Hydrogen Energy,2016,41(27):11616−11623. doi: 10.1016/j.ijhydene.2016.02.074
YAO L, WANG Y, SHI J, XU H L, SHEN W, HU C W. The influence of reduction temperature on the performance of ZrOx/Ni-MnOx/SiO2 catalyst for low-temperature CO2 reforming of methane[J]. Catal Today,2017,281(1):259−267.
ZHANG S H, MURATSUGU S, ISHIGURO N, TADA M. Ceria-doped Ni/SBA-16 catalysts for dry reforming of methane[J]. ACS Catal,2013,3(8):1855−1864. doi: 10.1021/cs400159w
LI K, PEI C L, LI X Y, CHEN S, ZHANG X H, LIU R, GONG J L. Dry reforming of methane over La2O2CO3-modified Ni/Al2O3 catalysts with moderate metal support interaction[J]. Appl Catal B Environ,2020,264:118448. doi: 10.1016/j.apcatb.2019.118448
KE Q, KANG L M, CHEN X, WU Y. DFT study of CO2 catalytic conversion by H2 over Ni13 cluster[J]. J Chem Sci,2020,132(1):151. doi: 10.1007/s12039-020-01857-3
YILMAZER N D, FELLAH M F, ONAL I. A DFT study of ethylene hydrogenation reaction mechanisms on Ni13 nanocluster[J]. Top Catal,2013,56(9/10):789−793. doi: 10.1007/s11244-013-0043-0
RUSINA G G, BORISOVA S D, CHULKOV E V. Structure and atomic vibrations in bimetallic Ni13-nAln clusters[J]. JETP Lett,2015,101(7):474−480. doi: 10.1134/S0021364015070139
BANERJEE R, DATTA S, MOOKERJEE A. Structure, reactivity and electronic properties of Mn doped Ni13 clusters[J]. Phys B,2013,419(21):86−89.
CHEN S J, CHEN X, ZHANG H. Probing the activity of Ni13, Cu13 and Ni12Cu clusters towards the ammonia decomposition reaction by density functional theory[J]. J Mater Sci,2016,52(6):3162−3168.
YILMAZER N D, FELLAH M F, ONAL I. A density functional theory study of ethylene adsorption on Ni10(111), Ni13(100) and Ni10(110) surface cluster models and Ni13 nanocluster[J]. Appl Surf Sci,2010,256(16):5088−5093. doi: 10.1016/j.apsusc.2010.03.067
YAO Y H, GU X, JI M, GONG X G, WANG D S. Structures and magnetic moments of Nin (n=10~60) clusters[J]. Phys Lett A,2007,360(4/5):629−631. doi: 10.1016/j.physleta.2006.08.059
LIU B, LUSK M T, ELY J F. Influence of nickel catalyst geometry on the dissociation barriers of H2 and CH4: Ni13 versus Ni(111)[J]. J Phys Chem C,2009,113(31):13715−13722. doi: 10.1021/jp9003196
GRIGORYAN V G, SPRINGBORG M. A theoretical study of the structure of Ni clusters (NiN)[J]. Phys Chem Chem Phys,2001,3(23):5135−5139. doi: 10.1039/b105831m
MORTENSEN J J, HANSEN L B, JACOBSEN K W. Real-space grid implementation of the projector augmented wave method[J]. Phys Rev B,2005,71:035109. doi: 10.1103/PhysRevB.71.035109
REXER E F, JELLINEK J, KRISSINEL E B, PARK E K, RILEY S J. Theoretical and experimental studies of the structures of 12-, 13-, and 14-atom bimetallic nickel/aluminum clusters[J]. J Chem Phys,2002,117(1):82−94. doi: 10.1063/1.1481386
STEFANOVICH E V, SHLUGER A L, CATLOW C R A. Theoretical study of the stabilization of cubic-phase ZrO2 by impurities[J]. Phys Rev B,1994,49(17):11560−11571. doi: 10.1103/PhysRevB.49.11560
EREMEEV S V, NEMIROVICH-DANCHENKO L Y, KUL′KOVA S E. Effect of oxygen vacancies on adhesion at the Nb/Al2O3 and Ni/ZrO2 interfaces[J]. Phys Solid State,2008,50:543−552. doi: 10.1134/S1063783408030256
BELTRÁN J I, GALLEGO S, CERDÁJ, MOYA J S, MUÑOZ M C. Bond formation at the Ni/ZrO2 interface[J]. Phys Rev B,2003,68(7):075401. doi: 10.1103/PhysRevB.68.075401
ZHANG M, ZIJLSTRA B, FILOT IAW, LI F, WANG HO, LI J D, HENSEN EJM. A theoretical study of the reverse water-gas shift reaction on Ni(111) and Ni(311) surfaces[J]. Can J Chem Eng,2020,98(3):740−748. doi: 10.1002/cjce.23655
MONKHORST H J, PACK J D. Special points for brillouin-zone integrations[J]. Phys Rev B,1976,13(12):5188−5192. doi: 10.1103/PhysRevB.13.5188
ZHI C M, YANG W. Improvement of Mo-doping on Sulfur-poisoning of Ni catalyst: Activity and selectivity to CO methanation[J]. Comput Theor Chem,2021,1197:113140. doi: 10.1016/j.comptc.2020.113140
KAPUR N, HYUN J, SHAN B, NICHOLAS J B, CHO K. Ab initio study of CO hydrogenation to oxygenates on reduced Rh terraces and stepped surfaces[J]. J Phys Chem C,2010,114(22):10171−10182. doi: 10.1021/jp911903u
AN W, CHEN X C, TURNER C H. First-principles study of methane dehydrogenation on a bimetallic Cu/Ni(111) surface[J]. J Chem Phys,2009,131(17):174702. doi: 10.1063/1.3254383
OU Z L, RAN J Y, NIU J T, ZHANG Z H, DENG T, HE Z Q, QIN C L. Effect of active site and charge transfer on methane dehydrogenation over different Co doped Ni surfaces by density functional theory[J]. Int J Hydrogen Energy,2020,45(56):31849−31862. doi: 10.1016/j.ijhydene.2020.08.187
PETERSSON G A, TENSFELDT T G, MONTGOMERY JR J A. Vinylidene and the hammond postulate[J]. J Am Chem Soc,1992,114:6133−6138. doi: 10.1021/ja00041a034
CHEN Z X, ALEKSANDROV H A, BASARAN D, RÖSCH N. Transformations of ethylene on the Pd(111) surface: A density functional study[J]. J Phys Chem C,2010,114(41):17683−17692. doi: 10.1021/jp104949w
ZHAO Z J, CHIU C C, GONG J L. Molecular understandings on the activation of light hydrocarbons over heterogeneous catalysts[J]. Chem Sci,2015,6(8):4403−4425. doi: 10.1039/C5SC01227A
KHETTAL H, HAROUN M F, BOUKELKOUL M. Theoretical study of CH4 adsorption and dissociation on W-Cu(100) surface[J]. Comput Theor Chem,2020,1186:112890. doi: 10.1016/j.comptc.2020.112890

Figure 2 Most stable configurations of adsorbed species involved in CH4 dissociation on Ni13 and Ni13-ZrO2(111), respectivelyThe blue, gray, red, white and turquoise balls represent Ni, C, O, H and Zr atoms, respectively Bond length are in Å

Figure 3 (a) Differential charge density, and (b) projected density of states (pDOS) for CH on Ni13 and Ni13-ZrO2(111)

Figure 4 Structures of the ISs, TSs and FSs relevant to CH4 dissociation on (a) Ni13 and (b) Ni13-ZrO2(111) Bond lengths are in Å see Figure 2 for color coding

Figure 5 Potential energy profile of C2 formations and C eliminations on (a) Ni13 and (b) Ni13-ZrO2(111) together with the corresponding structures
Bond lengths are in Å see Figure 2 for color coding

Figure 6 Partial density of states for CH3 adsorbed on Ni13 and Ni13-ZrO2(111) for the role of the support ZrO2: (a) broadening d-band, (b) stabilizing Ni−C bond and (c) activating C−H bond
Table 1. Adsorption sites and adsorption energies (Eads) of the stable configurations for the adsorbed species involved in CH4 dissociation on Ni13 and Ni13-ZrO2(111), respectively
| Species | Ni13 | Previous results Ni4[6], Ni(100)[7] | Ni13-ZrO2(111) | |||
| site | Eads/eV | Eads/eV | site | Eads/eV | ||
| CH4 | Ni-top | 0.04 | −0.41[6] | Ni-top | −0.02 | |
| CH3 | Ni-bridge | −2.22 | −2.37[6] | Ni-bridge | −2.46 | |
| CH2 | Ni-fold | −4.36 | −4.85[6], −3.76[7] | Ni-fold | −4.76 | |
| CH | Ni-fold | −6.40 | −6.71[6], −6.43[7] | Ni-fold | −7.00 | |
| C | Ni-fold | −7.21 | −8.94 [6], −7.27[7] | Ni-fold | −7.96 | |
| H | Ni-fold | −2.78 | −2.41[6], −2.36[7] | Ni-fold | −3.02 | |
| DFT methods: Gaussian 09W code, GGA-PBE[6]; ADF-BAND code, GGA-RPBE[7] | ||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Activation energies the reaction energies the C−H bonds length of ISs and TSs (dC–H/Å) and H displacements (Å) involved in CH4 dissociation on Ni13 and Ni13-ZrO2(111)
| Reaction | Our results | Previous results | ||||||||
| Ea/eV | ΔE/eV | v/cm−1 | dC–H ISs/Å | dC–H TSs/Å | H displacements/Å | Ea/eV | ΔE/eV | |||
| Ni13 | Ni4[6] | Ni(100)[7] | ||||||||
| CH4→CH3+H | R1-1 | 0.61 | −0.50 | 932 i | 1.089 | 1.561 | 0.472 | 1.23[7] | ||
| CH3→CH2+H | R1-2 | 0.46 | −0.15 | 416 i | 1.107 | 2.337 | 1.230 | 0.84[6], 0.62[7] | −1.41[6] | |
| CH2→CH+H | R1-3 | 0.48 | −0.22 | 625 i | 1.107 | 1.795 | 0.688 | 0.33[6], 0.22[7] | −4.40[6] | |
| CH→C+H | R1-4 | 0.70 | −0.07 | 747 i | 1.107 | 1.479 | 0.372 | 1.37[6], 0.64[7] | −5.96[6] | |
| C+C→C2 | R1-5 | 3.80 | 0.62 | 524 i | ||||||
| Ni13-ZrO2(111) | ||||||||||
| CH4→CH3+H | R2-1 | 0.14 | −0.66 | 59 i | 1.104 | 1.122 | 0.018 | |||
| CH3→CH2+H | R2-2 | 0.35 | −0.47 | 760 i | 1.143 | 1.662 | 0.519 | |||
| CH2→CH+H | R2-3 | 0.14 | −0.53 | 488 i | 1.189 | 1.782 | 0.593 | |||
| CH→C+H | R2-4 | 0.40 | −0.30 | 657 i | 1.108 | 1.389 | 0.281 | |||
| C+C→C2 | R2-5 | 1.90 | 1.87 | 170 i | ||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

