
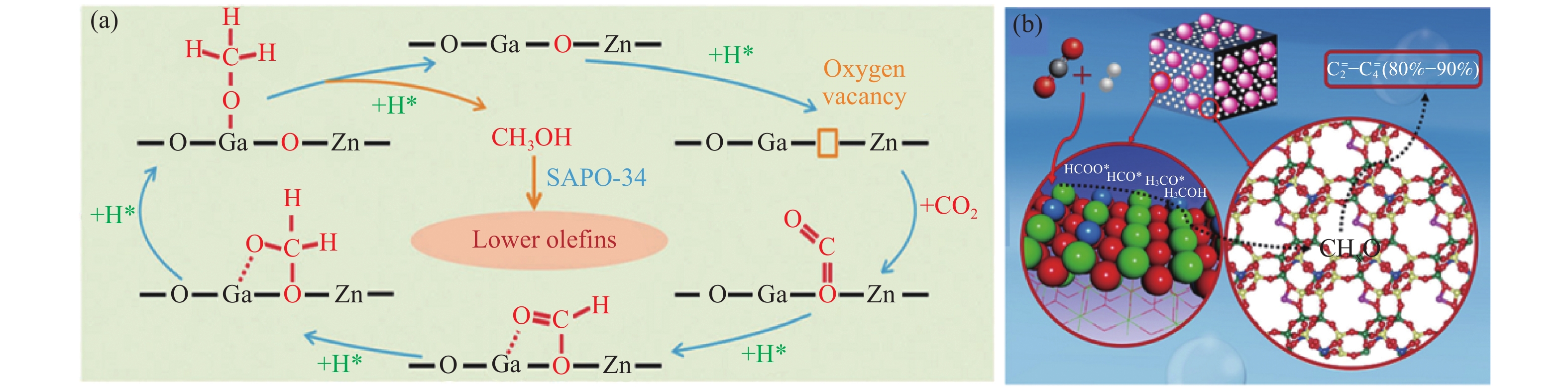
图 1.
CO2加氢制备C2+烃的串联催化过程
Figure 1.
Tandem catalysis of CO2 hydrogenation into C2+ hydrocarbons

当前,能源和环境问题引起了全社会的广泛关注,是限制和影响人类未来发展的两大核心问题。随着中国现代工业和城市化进程的快速发展,造成了CO2排放量的剧烈增加,且中国短时间内以煤炭和石油为主的能源结构难以改变。CO2的大量排放,不仅污染环境,而且引起碳资源的巨大浪费。为了应对全球气候变化和环境污染带来的挑战,实现中国经济的可持续发展,2020年习总书记向全世界作出郑重承诺,“力争于2030年前二氧化碳排放达到峰值,2060年前实现碳中和”。除了提高能源使用效率以及对CO2进行捕获和封存等手段降低CO2排放外,积极发展CO2催化转化技术是兼顾经济发展和遏制温室效应的重要途径。而且,从长远来看,将CO2催化转化为高附加值化学品或液体燃料比CO2捕获及封存更有意义,能够有效实现碳资源的高值化及循环利用。
尽管CO2是一种廉价、无毒、丰富易得的可再生碳资源,但CO2的化学结构高度稳定(ΔGf0 = –394.4 kJ/mol),CO2中碳原子处于最高的氧化状态,同时其动力学惰性,极其不活泼,将其转化为高附加值化学品时需要较高的能量输入[1,2]。因此,实现CO2催化转化需要添加高能量的还原剂打破C–O键(例如H2),或者提供额外的能量辅助(例如:电能或光能)。虽然电催化和光催化目前也受到研究人员的广泛关注,但从工业放大角度考虑,热催化仍然是CO2催化加氢制备高附加值化学品及液体燃料最有希望的合成路线,而最值得担心的H2来源问题,可通过可再生能源(例如风能、生物质或太阳能等)发电及电解水制得。
CO2加氢可催化转化生成甲醇[3-10]、乙醇[11-13]、二甲醚[14-17]、LPG[18,19]及低碳烃[20-23]等高附加值的化学品。然而,高效催化剂的缺乏导致CO2加氢活性低及C–C键偶联可控程度差,因此,CO2加氢制备较高碳数烃的研究受到明显抑制。近年来,随着Fe基或金属氧化物与分子筛串联催化剂的出现,CO2加氢转化制备高附加值化学品及液体燃料取得了突破性的研究进展,CO2加氢可分别通过甲醇或CO中间体反应路径高选择性地生成C2+烃(主要包括低碳烯烃、异构烷烃、汽油及芳烃等)[24-28]。串联催化剂中两类活性组分间不仅需要良好匹配,而且活性组分的组装方式、分子筛的性质及反应参数均对烃类产物的生成影响显著。目前,针对串联催化剂上CO2催化转化制备C2+烃类的综述文章并不多见。因此,本综述针对目前报道的CO2加氢制备低碳烯烃、异构烷烃、汽油及芳烃等串联催化过程,重点阐述串联催化剂上影响CO2转化及目标产物生成的关键因素以及串联催化剂的稳定性,同时对CO2加氢串联催化过程的未来发展进行展望。
早在二十世纪末,“串联催化(Tandem catalysis)”概念就已在合成化学中出现[29,30],目前,串联催化在许多反应体系中也得到了应用[31,32]。串联催化不仅可以通过活性组分间的有效耦合拉动反应的进行,而且还能省去中间产物的净化分离过程,降低反应能耗。近年来,串联催化在合成气及CO2加氢转化制备高附加值化学品及液体燃料方面均得到了广泛应用,且已取得突破性的研究进展[24-28,33-38]。Li等[39,40]将串联催化策略应用于CO2加氢反应过程,金属氧化物与分子筛间的良好匹配促使CO2通过加氢高选择性转化为低碳烯烃或芳烃。尽管串联催化在CO2加氢过程明确提出较晚,但串联催化剂同样在二十世纪末已被用于CO2加氢制备低碳烃反应,不同文献对两类活性组分形成的复合催化剂命名不同,例如串联催化剂[39,40]、双功能催化剂[33,34,36,38,41-44]、核壳催化剂[26,45-47]、接力催化剂[2]等。
图1所示为CO2加氢制备C2+烃的串联反应过程示意图。CO2及H2首先在Fe基催化剂(Fe)或金属氧化物(OX)活性中心上吸附、活化及反应生成中间产物,生成的中间产物通过分子筛的孔道转移到酸中心上,再经连续的酸催化过程生成目标产物C2+烃。截止目前,CO2加氢制备C2+烃的研究报道主要集中于低碳烯烃、异构烷烃、汽油及芳烃等方面。串联催化反应除了需要不同的活性组分外,还必须考虑以下两方面因素:一是,反应条件的匹配,由于串联催化在同一个反应装置中进行,因此不同反应过程的操作条件必须匹配,才能保证反应顺利进行;二是, 活性组分间的协同匹配,串联催化剂中每种活性组分都必须与反应物、中间物种及最终产物相互兼容,而且活性组分间也必须高效协同,才能更有利于目标产物的生成。
传统地,CO2催化加氢制备碳氢化合物反应主要利用改性费托合成催化剂。该反应在相同金属活性相上发生CO2活化和C−C键生长,即CO2还原为CO,随后CO通过费托合成反应生成碳氢化合物。由于费托合成反应中产物碳链生长遵循聚合机理,以链式碳氢化合物为主,因此,产物分布宽,导致产品组成较为复杂。从反应机理上,费托合成反应更难以生成分子结构复杂的碳氢化合物,如芳烃和环烷烃。近些年,串联催化策略在CO2加氢制低碳烯烃、异构烷烃、汽油及芳烃反应中得到了广泛应用。目前,CO2加氢制低碳烯烃反应主要采用金属氧化物与分子筛串联催化剂,遵循甲醇中间体的反应路径。然而,对于CO2加氢制异构烷烃、汽油及芳烃的反应体系则分别采用Fe/zeolite或OX/zeolite串联催化剂,在这两类串联催化剂上CO2加氢分别遵循CO中间体的FTS反应路径或甲醇中间体反应路径。
低碳烯烃(
在氧化物与分子筛串联体系中,高效的甲醇合成氧化物以及氧化物与分子筛间的良好协同是高选择性合成低碳烯烃的关键。表1总结了具有代表性的金属氧化物与分子筛串联催化剂上CO2加氢制备低碳烯烃的催化性能。可还原的In2O3由于其表面存在丰富的氧空穴,能够有效的活化CO2分子,被广泛用于CO2加氢制甲醇[59-62]或经甲醇中间体制低碳烯烃反应中[24,25,41]。Gao等[24]发现,共沉淀法制备的In-Zr双金属氧化物与SAPO-34分子筛复合形成的串联催化剂表现出优异的CO2加氢制低碳烯烃性能。在400 ℃,3 MPa,9000 mL/(gcat·h)条件下,CO2的转化率及
 下载:
导出CSV
下载:
导出CSV
| Catalyst | t /℃ | p /MPa |   |   |
| In-Zr/SAPO-34 (Granule-mixing)[24] | 400 | 3 | 35.5 | 76.4 |
| In2O3-ZnZrOx/SAPO-34 (Granule-mixing)[41] | 380 | 3 | 17.0 | 85.0 |
| 1In2O3/ZrO2-1SAPO-34 (Powder-mixing)[42] | 400 | 1.5 | ~20.0 | 80.0–90.0 |
| InCrOx(0.13)/SAPO-34 (Powder-mixing)[25] | 350 | 1 | 17.2 | 89.1 |
| ZnGa2O4/SAPO-34 (Powder-mixing)[43] | 370 | 3 | 13.0 | 86.0 |
| ZnZrO/SAPO-34 (Powder-mixing)[39] | 380 | 2 | 12.6 | 80.0 |
| Zr8Cd1/SAPO-18 (Powder-mixing)[44] | 370 | 2.5 | 17.8 | 85.6 |
| CuZnZr@Zn–SAPO-34 (Core-shell)[45] | 400 | 2 | 19.6 | 60.5 |
| Zn0.5Ce0.2Zr1.8O4/H-RUB-13 (Powder-mixing)[57] | 350 | 1 | 10.7 | 83.4 |
| Mn2O3-ZnO/SAPO-34 (Ball milling-mixing)[58] | 380 | 3 | 29.8 | 80.2 |
*:     | ||||
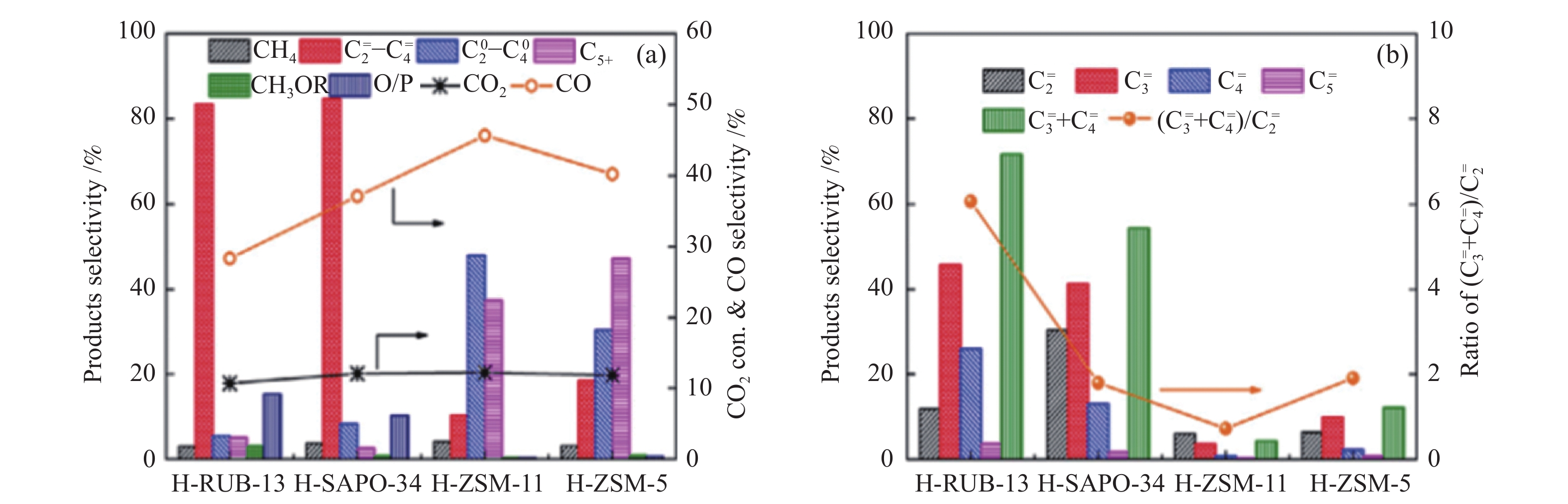
此外,Wang等[25]发现,将适当的Ce或Cr离子引入到In2O3晶格中获得的InCeOx(0.13)及InCrOx(0.13)氧化物上CO2加氢时,在300 ℃,1.0 MPa,4800 mL/(gcat·h)条件下,甲醇的选择性明显高于单纯的In2O3,同时甲烷的选择性显著降低。实验及DFT理论计算表明,InCeOx(0.13)及InCrOx(0.13)均产生更多的表面氧空穴,而且提高了HCOO*物种与催化剂表面的电子相互作用。与SAPO-34分子筛复合形成的InCeOx(0.13)/SAPO-34及InCrOx(0.13)/SAPO-34双功能催化剂上CO2加氢时,在300 ℃,1.0 MPa,4000 mL/(gcat·h)条件下,
除上述In基氧化物与SAPO-34分子筛复合催化剂被用于CO2加氢制低碳烯烃外,一些其他的甲醇合成催化剂(ZnGa2O4、ZnZrO或Zr8Cd1等)也与SAPO-34分子筛耦合用于该串联催化过程(表1)。Liu等[43]考察了尖晶石结构的ZnGa2O4氧化物与SAPO-34分子筛串联催化剂上CO2加氢制低碳烯烃的性能。研究发现,ZnGa2O4表面的氧空穴是CO2活化及通过甲酸盐HCOO*及甲氧基CH3O*物种形成甲醇的活性中心(图2(a))。在370 ℃,3 MPa,5400 mL/(gcat·h)条件下,CO2的转化率及烃中
(a): Zn-Ga-O/SAPO-34[43]; (b): ZnZrO/SAPO[39] reproduced with permission from ref.[43, 39], Copyright (2018) The Royal Society of Chemistry, Copyright (2017) American Chemical Society
此外,Cu-Zn基复合氧化物作为典型的甲醇合成催化剂,被广泛应用于CO2加氢制甲醇反应[57,63,64]。然而,Cu基氧化物及分子筛串联催化剂很少被用于CO2加氢经甲醇中间体制备低碳烯烃的反应中[43,65,66]。Liu等[43]发现,Cu-Zn-Al/SAPO-34串联催化剂上CO2加氢制烯烃,反应温度<300 ℃时,Cu-Zn-Al上生成的甲醇及二甲醚并未在分子筛上完全转化生成烯烃;当反应温度≥350 ℃时,烃产物中
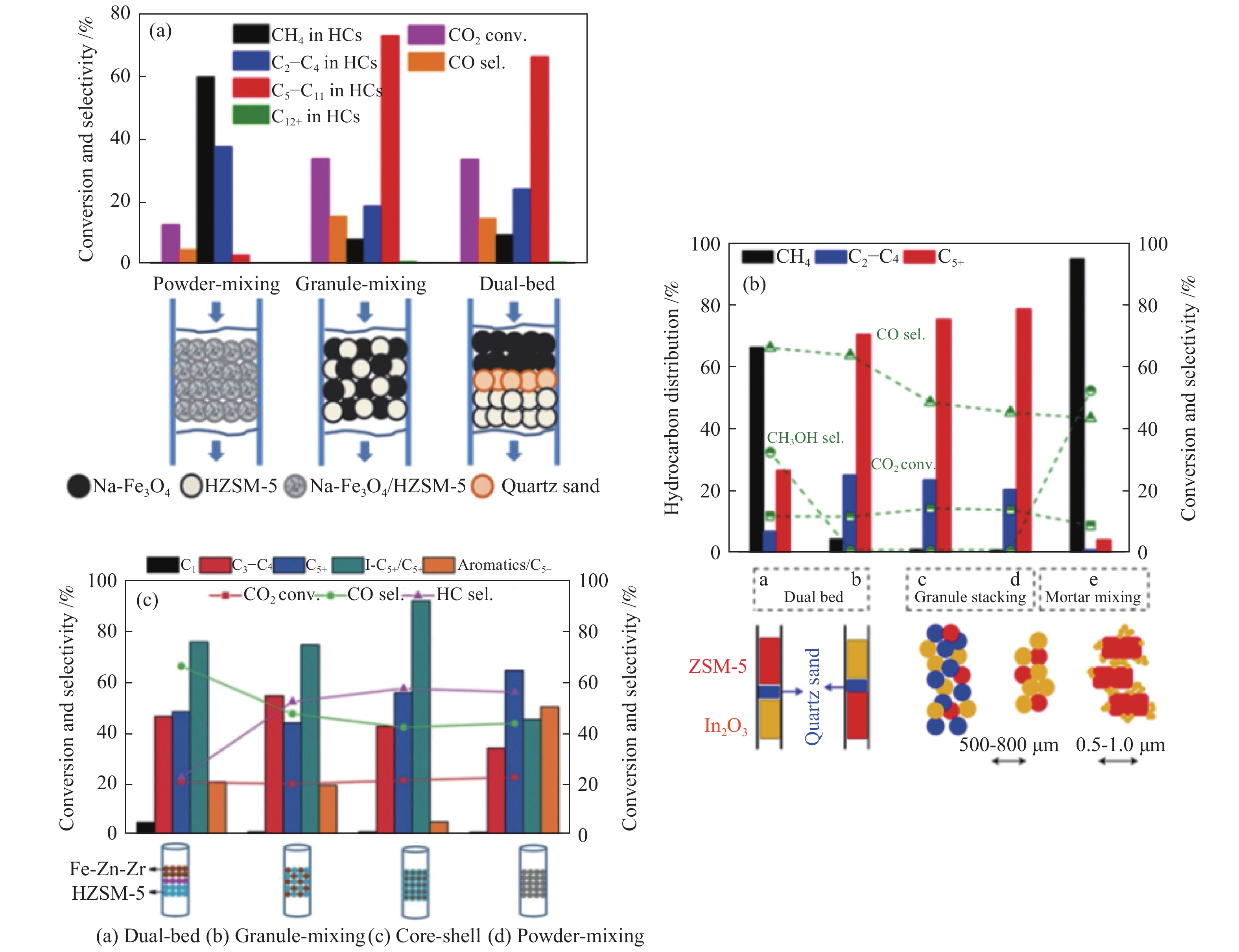
对于串联催化剂来说,两类活性组分间不仅需要良好的协同作用,而且两种组分间的组装方式对复合催化剂的性能同样影响较大。由表1可知,尽管不同的金属氧化物与SAPO-34分子筛耦合形成的串联催化剂上,CO2加氢均可高选择性的合成低碳烯烃,但金属氧化物与分子筛间的组装方式并不相同,这可能归因于氧化物自身性质的差异。也就是说,氧化物的性质决定了其与分子筛串联时,两类活性组分间最佳的结合方式,即两类活性组分间的接近度。如图3(a)所示,In-Zr氧化物与SAPO-34分子筛分别以双床层、颗粒混合及粉末方式结合时,串联催化剂上烃类产物的选择性明显不同[24]。当以双床层方式结合时,两类活性组分间的距离最远,烃产物中CH4选择性高达70%,
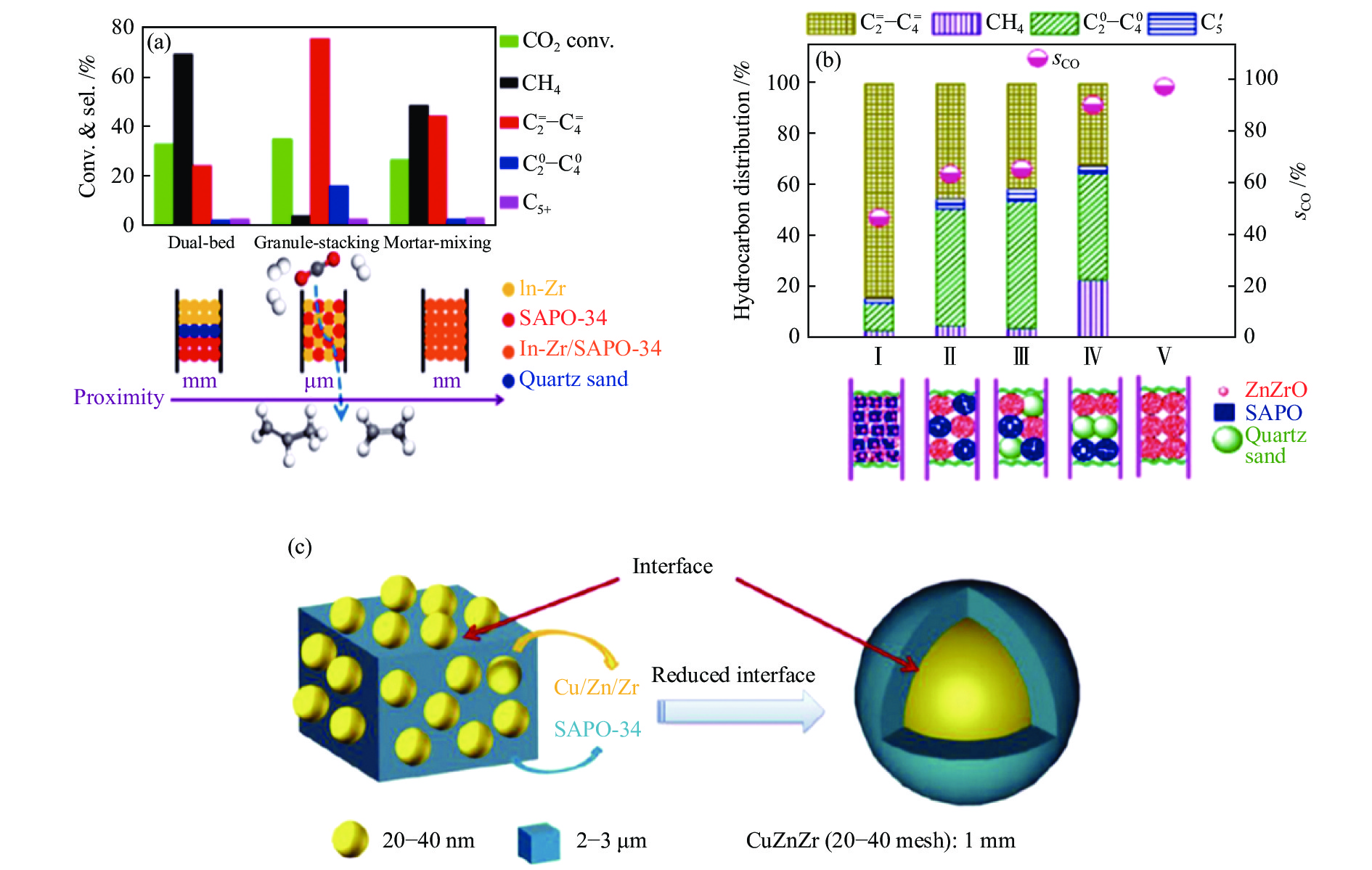
与此同时,Gao等[42]通过沉积沉淀法将In2O3负载于ZrO2载体上(In∶Zr = 1∶10.7(物质的量比))制备的1In2O3/ZrO2-1SAPO-34串联催化剂上CO2加氢制低碳烯烃时,在400 ℃,1.5 MPa,12000 mL/(gcat·h)条件下,CO2的转化率及低碳烯烃(乙烯和丙烯)的选择性分别为~20%及80%−90%,CO的选择性>80%。在不同反应温度下,In2O3/ZrO2与SAPO-34以粉末方式混合时,总烃及烯烃的选择性均明显高于颗粒混合时两者的选择性。然而,上述Gao等[24]报道的共沉淀法制备的In-Zr氧化物(In∶Zr = 1∶4(物质的量比))与SAPO-34分子筛以颗粒方式混合时,低碳烯烃的选择性最高。同样是In-Zr氧化物与SAPO-34分子筛组分,催化性能最佳时两组分的结合方式却明显不同,这可能因为In-Zr氧化物不同的制备方法及In∶Zr比例导致两者具有不同的物化性质,引起不同的CO2活化及加氢能力,从而它们与分子筛耦合时最佳的结合方式有所不同。而且,In-Zr/SAPO-34(Granule-mixing)[24]与1In2O3/ZrO2-1SAPO-34(Powder-mixing)[42]两催化剂上CO2转化率相差近一倍,尽管反应条件存在一定差异,但在一定程度上也表明了两种In-Zr氧化物具有不同的CO2活化能力(表1);另一方面,同样是与SAPO-34分子筛混合,两种In-Zr氧化物在反应过程中In发生迁移的程度也可能有所不同,从而导致活性组分间最佳结合方式存在差异。
此外,具有尖晶石结构的ZnGa2O4氧化物或ZnZrO固溶体与SAPO-34分子筛复合形成的串联催化剂上,CO2加氢制低碳烯烃最优的反应性能同样并非为两类活性组分以颗粒方式混合。Wang等[43]报道的ZnGa2O4与SAPO-34粉末混合的串联催化剂上CO2加氢时,在370 ℃,3 MPa,5400 mL/(gcat·h)条件下,CO2的转化率及烃中
如前文所述,Cu-Zn-Al/SAPO-34串联催化剂[43]上CO2加氢制烯烃时,由于Cu物种非常强的加氢性能,导致烃类产物中烯烃极易加氢生成烷烃。近来,Chen等[45]研究了CZZ@Zn-SAPO-34核壳催化剂上CO2加氢制低碳烯烃的性能。在400 ℃,2 MPa,6000 h−1反应条件下,CZZ@Zn-SAPO-34上低碳烯烃选择性可高达72%,明显高于CZZ/Zn-SAPO-34粉末混合催化剂上低碳烯烃的选择性。与粉末混合催化剂相比,核壳催化剂上烯烃选择性提高的原因主要为氧化物与分子筛间的接触界面减小及甲醇中间体的浓度降低(图3(c)),从而导致催化剂上进行烯烃加氢及氢转移反应的几率降低;另一方面,SAPO-34分子筛上Zn物种的引入导致其酸性降低,从而抑制二次加氢反应的进行。与Li等[39]报道的ZnZrO/SAPO串联催化剂不同,ZnZrO及SAPO以粉末方式混合时,
因此,对于CO2加氢制低碳烯烃反应来说,由于不同金属氧化物的活性及加氢能力的差异,导致其与分子筛耦合时,两组分间的最佳结合方式明显不同。当氧化物活性较高时,氧化物与分子筛间的距离不能太过接近,太近的距离会导致低碳烯烃进一步加氢生成烷烃,降低目标产物的选择性;反之,当氧化物活性较低时,两组分需要充分接触来提高中间产物的进一步转化,即以最近的接近度(粉末方式混合)复合时,目标产物
由于氧化物与分子筛串联催化剂上CO2加氢经甲醇或甲醇前驱物种等进一步在分子筛酸中心上转化生成低碳烯烃,因此,除氧化物自身性质对CO2转化及低碳烯烃生成具有显著影响外,分子筛的结构及酸性对低碳烯烃的选择性同样具有显著影响。Dang等[41]发现,In2O3-ZnZrOx/SAPO-34串联催化剂上CO2加氢制低碳烯烃时,随SAPO-34分子筛晶粒尺寸的减小,
此外,Wang等[67]研究了不同孔道结构的分子筛对低碳烯烃选择性的影响,如图4所示。Zn0.5Ce0.2Zr1.8O4固溶体分别与SAPO-34分子筛及与SAPO-34分子筛结构类似的H-RUB-13分子筛串联时,
Gao等[24]报道的In-Zr/SAPO-34串联催化剂上CO2加氢制低碳烯烃时,随H2/CO2比例及反应压力的增加,CO2的转化率及
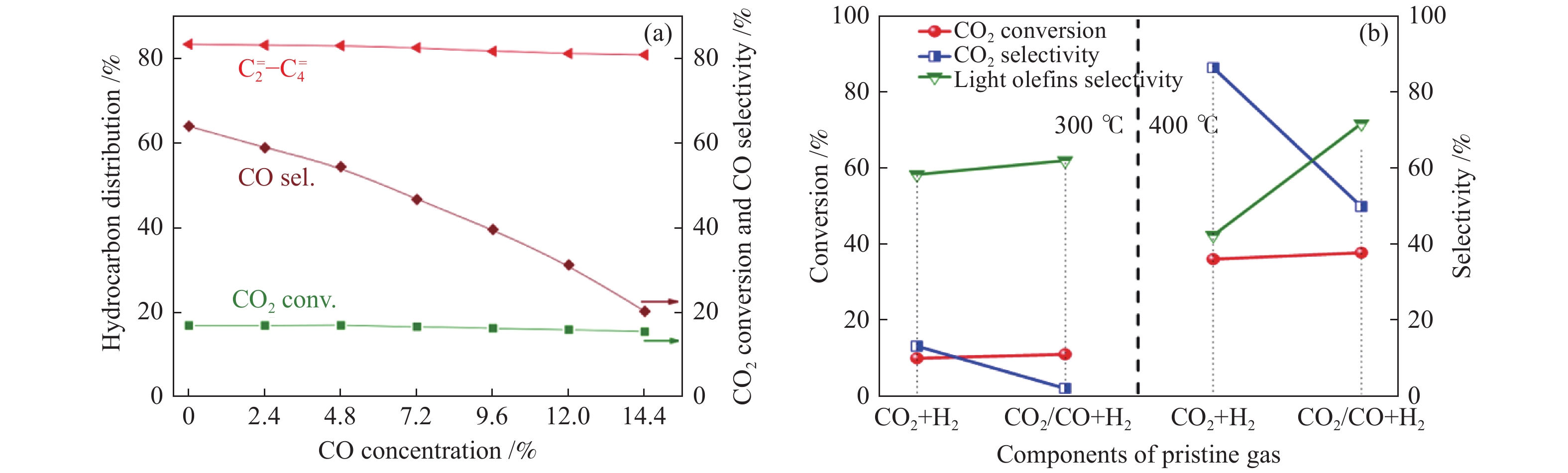
除改变反应条件抑制副产物CO的生成外,向原料气中加入CO也可显著抑制逆水煤气反应[24]。如图5(a)所示,随原料气中CO比例增加至14.4%,CO的选择性降至20.5%,CO2的转化率及
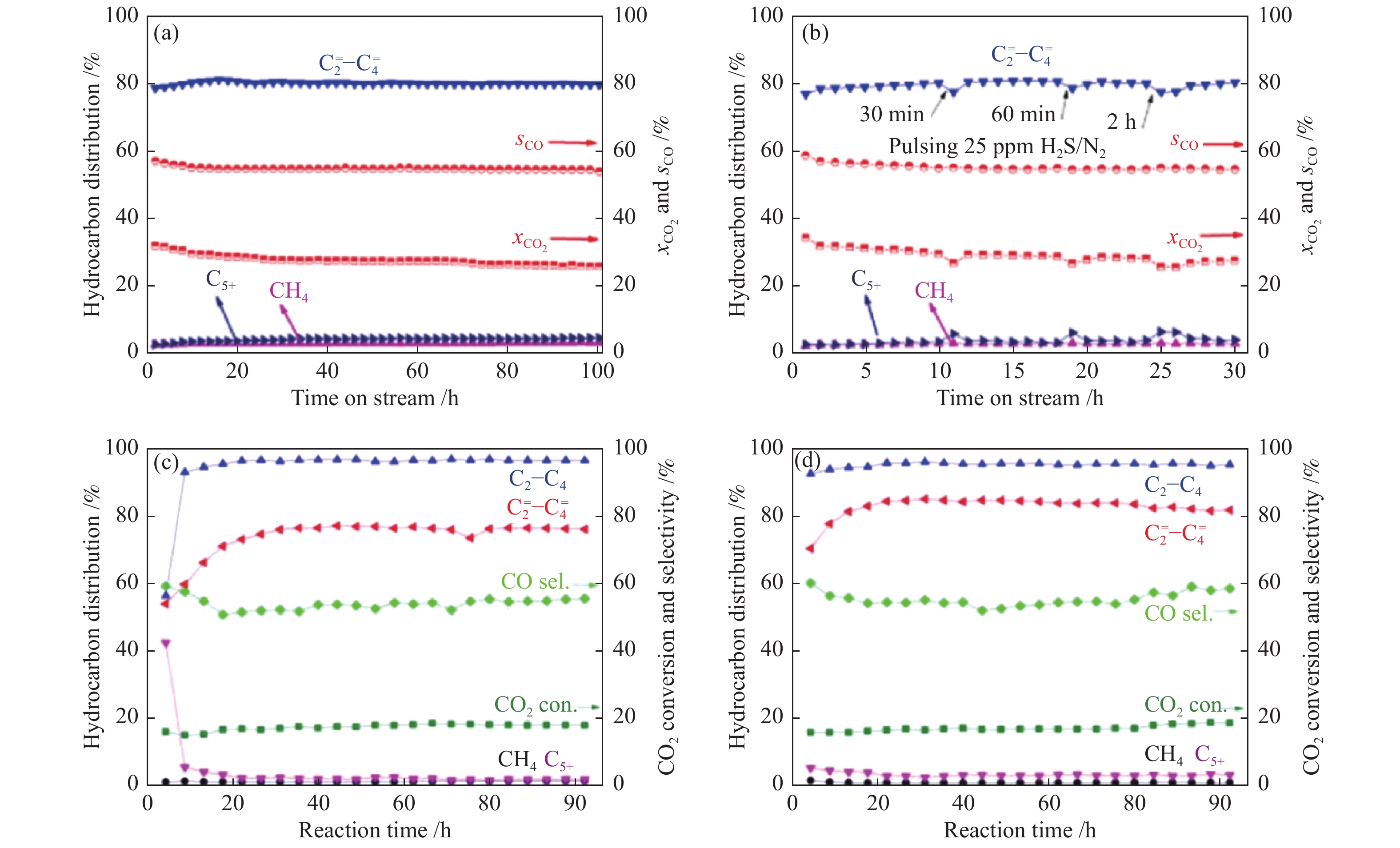
催化剂的稳定性及抗毒性是决定其能否在实际工业中广泛应用的关键因素,因此,也是串联催化剂需要关注的问题。Mou等[58]发现,20%Mn2O3-ZnO/SAPO-34串联催化剂在CO2加氢制低碳烯烃反应中具有非常好的稳定性及抗硫性能。如图6(a), (b)所示,该串联催化剂在380 ℃,5 MPa,3600 mL/(gcat·h)条件下连续运转100 h时,CO2的转化率及
此外,Dang等[41]考察了In2O3-ZnZrOx/SAPO-34-C及In2O3-ZnZrOx/SAPO-34-H-a串联催化剂上CO2加氢制低碳烯烃的稳定性。In2O3-ZnZrOx/SAPO-34-C串联催化剂连续运行92 h时,CO2转化率及
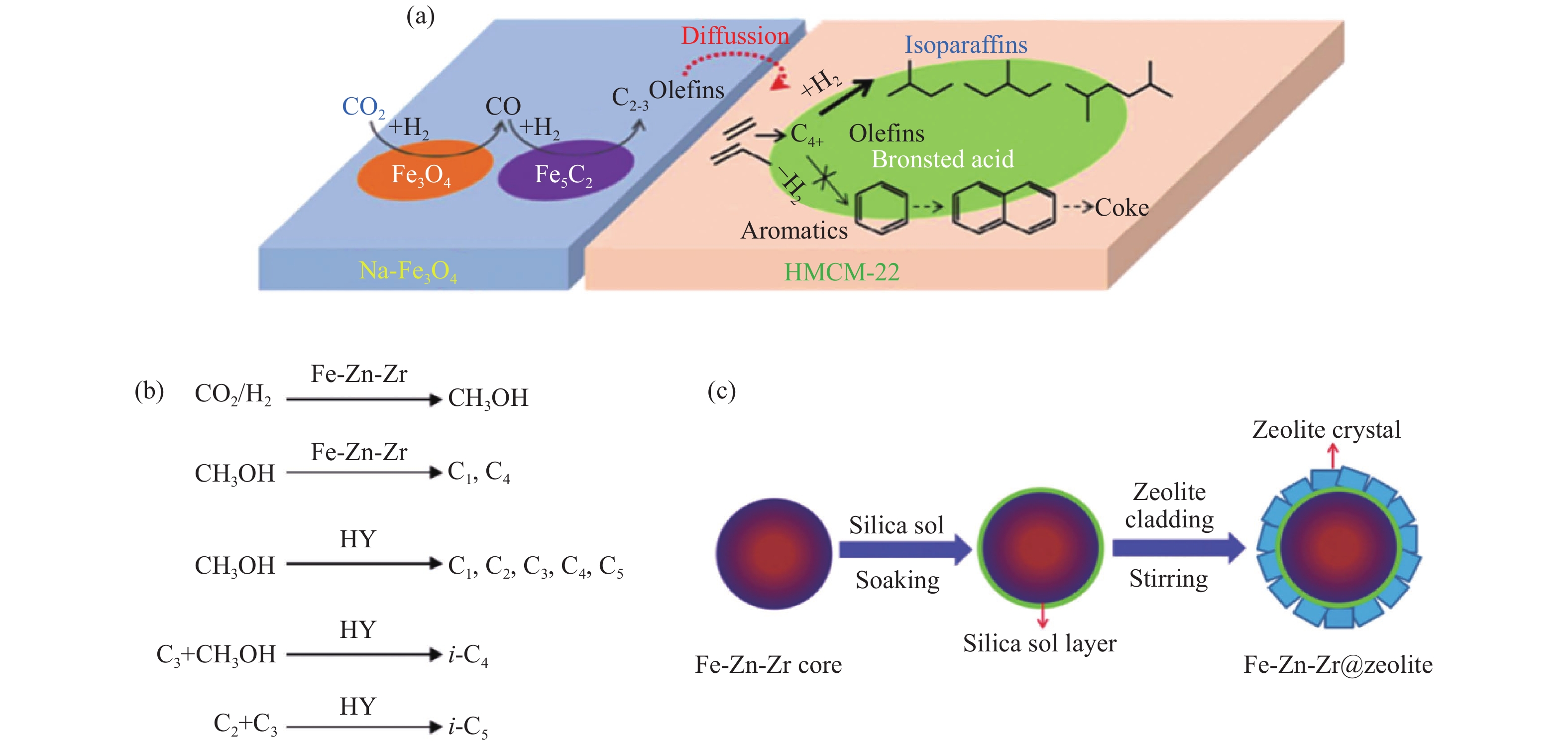
当CO2加氢制异构烷烃及汽油遵循CO中间体的FTS反应路径时,主要采用碱金属改性的Fe基与分子筛复合催化剂。首先,Xu等[70,71]较早报道了Fe-Cu-Na/zeolite 复合催化剂上CO2加氢的催化性能。研究表明,恰当Na含量的Fe-Cu-Na与分子筛物理混合时,复合催化剂的活性及支链烃和高碳烃的选择性均较单纯的Fe-Cu-Na氧化物明显提高。他们认为,固相氧化物与分子筛间会发生反应,氧化物上的Na离子向分子筛表面迁移,导致Fe物相的还原程度增加,从而提高催化剂的F-T活性。近年来,Wei等[72]报道Na-Fe3O4与HMCM-22分子筛串联催化剂上CO2加氢达到了更高的异构烷烃收率。结果表明,总烃中C4+烃的选择性达82%,C4+烃中异构烷烃选择性占74%(见表2),同时CO2的转化率及CO的选择性分别为26%及17%。这主要归因于Na-Fe3O4/HMCM-22复合催化剂实现了逆水煤气反应、C–C偶联及异构化反应间的良好匹配,同时HMCM-22分子筛合适的孔道结构及B酸中心有效促进烯烃聚合及异构化反应。Na-Fe3O4/HMCM-22催化剂上异构烷烃合成及积炭形成的反应机制如图7(a)所示。CO2首先在Fe3O4相上通过逆水煤气反应生成CO,生成的CO中间体再在Fe5C2相上通过FTS反应路径生成低碳烯烃,然后进一步扩散到分子筛的B酸中心上经聚合及异构化反应生成异构烷烃。然而,HMCM-22分子筛的孔道结构导致低碳烯烃易于发生芳构化反应生成稠环芳烃等积炭物种,但催化剂再生后性能保持良好。除Na离子外,K改性的Fe催化剂与分子筛混合制备的92.6Fe7.4K+HZSM-5也被用于CO2加氢制异构烷烃反应[73]。在300 ℃,2.5 MPa,560 mL/(gcat·h)反应条件下,CO2的转化率及C4–6烃中异构烷烃的选择性分别达43.9%及69.7%(表2)。
(a), (b): 20%Mn2O3-ZnO/SAPO-34[58]; (c): In2O3-ZnZrOx/SAPO-34-C[41]; (d): In2O3-ZnZrOx/SAPO-34-H-a[41] reproduced with permission from ref. [58, 41], Copyright (2021) Elsevier, Copyright (2019) John Wiley and Sons
对于不含碱金属离子的Fe-Zn基氧化物及分子筛复合催化剂上CO2加氢时,通常认为甲醇是主要的反应中间体[75–78]。Bai等[76]考察了Fe-Zn基氧化物与分子筛复合催化剂上CO2加氢制异构烷烃性能。结果表明,对于不同的金属助剂(Al、Mn、La、Cr、Zr)来说,Zr组分的效果最佳,Fe-Zn-Zr与HY分子筛复合催化剂有效耦合了甲醇合成及甲醇转化反应,在340 ℃,5.0 MPa,3000 mL/(gcat·h)条件下,CO2的转化率及总烃中异构烷烃的选择性分别达22.4%及55.3%(表2)。随后,Ni等[78]通过对比Fe-Zn-Zr及Fe-Zn-Zr/HY催化剂上CO2加氢及CO加氢的产物分布,提出一条CO2加氢制异构烷烃的反应路径(图7(b))。CO2首先通过加氢生成甲醇,然后甲醇通过MTG反应生成小分子烯烃
下载:
导出CSV
| Catalyst | t /℃ | p /MPa |   | sisoalkanes or gasoline /% |
| Fe-Cu-Na/US-Y(10.7) (Physical-mixing)[71] | 250 | 2 | 11.5 | 77.3a |
| Na-Fe3O4/HMCM-22(Dual-bed)[72] | 320 | 3 | 26.0 | 74.0b |
| 92.6Fe7.4K+HZSM-5 (Granule-mixing)[73] | 300 | 2.5 | 43.9 | 69.7a |
| NaFe+SAPO-11+HZSM-5(Triple-bed)[74] | 320 | 3 | 31.2 | 38.2c |
| Fe-Zn-Zr/HY (Granule-mixing)[76] | 340 | 5 | 22.4 | 55.3a |
| Fe-Zn-Zr@HZSM-5-Hbeta (Core-shell)[46] | 340 | 5 | 14.9 | 81.3d |
| Fe-Zn-Zr@HZSM-5 (Core-shell)[47] | 340 | 5 | 21.5 | 91.9e |
| Na-Fe3O4/HZSM-5 (Granule-mixing)[79] | 320 | 3 | 22.0 | 78.0f |
| In2O3/HZSM-5 (Granule-mixing)[80] | 340 | 3 | 13.1 | 78.6f |
| a: C4–C6 isoalkanes selectivity in C4–C6 hydrocarbons;b: C4+ isoalkanes selectivity in C4+ hydrocarbons;c: C5+ isoalkanes selectivity in all hydrocarbons;d: C4+ isoalkanes selectivity in all hydrocarbons;e: C5+ isoalkanes selectivity in C5+ hydrocarbons;f: C5–C11 or C5+ hydrocarbons in all hydrocarbons | ||||
此外,Wei等[79]发现,Na-Fe3O4与HZSM-5复合催化剂上,CO2加氢遵循CO中间体的FTS反应路径,Fe3O4、Fe5C2及B酸中心三者间良好的匹配促使CO2加氢高选择性转化为汽油组分,CO2转化率为22%,总烃中C5−C11汽油烃的选择性高达78%(表2),甲烷的选择性仅4%;而且,Na–Fe3O4/HZSM-5串联催化剂具有良好的稳定性,连续反应1000 h,C5+烃的选择性仍可保持在(67±2)%。与此同时,孙予罕课题组的Gao等[80]发现,In2O3/HZSM-5复合催化剂上CO2加氢制汽油时,总烃中汽油烃的选择性达78.6%。在此复合催化剂上,由于In2O3表面具有的丰富氧空穴,可有效的活化CO2及H2形成甲醇中间物种,然后通过分子筛的酸中心经甲醇转化路径进一步转化生成汽油烃。
无论对于哪种反应路径的串联催化剂来说,两类活性组分间的组装方式均对催化性能具有显著影响。Na-Fe3O4与HZSM-5分子筛间的组装方式对串联催化剂的性能影响如图8(a)所示。与颗粒混合及双床层混合相比,两组分以粉末混合时,CO2的转化率明显较低,烃产物中不期望产物CH4的选择性高达60%,说明Na-Fe3O4及HZSM-5两类活性组分间以最近的距离结合时并不利于汽油范围烃(C5–C11)的生成。然而,Na-Fe3O4及HZSM-5以颗粒方式混合时,C5–C11的选择性达73%。同时,对两组分颗粒混合或双床层的烃产物分布详细对比发现,颗粒混合更有利于芳烃生成,而双床层有利于生成异构烷烃。随后,Noreen等[74]进一步研究了Na-Fe3O4与SAPO-11及HZSM-5双分子筛串联催化剂催化CO2加氢制备多支链烃。结果表明,三组分以三床层装填方式结合时,烃产物中多支链烃的选择性明显提高,CO2转化率为31.2%时,汽油烃及汽油范围异构烷烃的选择性分别达71.7%及38.2%,辛烷值RON达91.7。
(a): Na-Fe3O4/HZSM-5[79]; (b): In2O3/HZSM-5[80]; (c): Fe-Zn-Zr@HZSM-5[47] reproduced with permission from ref. [79, 80, 47], Copyright (2017) Springer Nature, Copyright (2019) The Royal Society of Chemistry
另外,In2O3及HZSM-5两组分间的接近度对抑制不期望的逆水煤气反应及选择性生成汽油烃同样具有重要作用。如图8(b)所示,In2O3及HZSM-5直接以颗粒方式混合或石英砂稀释时,CO2的转化率及烃产物中C5+烃的选择性均较高,同时副产物CO的选择性较低;然而,增加两类活性组分间的距离,以双床层方式装填时,C5+烃的选择性有所降低,同时副产物CO明显增加。而且,与Na改性的Fe基催化剂类似,In2O3及HZSM-5以粉末方式混合时,CH4的选择性急剧增加至94.3%,C5+烃的选择性仅4.2%,同时发现中间产物甲醇的选择性显著增加(51.9%),这可能归因于HZSM-5分子筛的失活导致甲醇不能完全转化。
Wang等[47]也考察了Fe-Zn-Zr氧化物与HZSM-5分子筛间的组装方式对CO2加氢制汽油组分的影响。Fe-Zn-Zr及HZSM-5两活性组分间的距离对烃类产物分布影响较大(图8(c))。当Fe-Zn-Zr与HZSM-5以核壳方式结合时,最有利于C5+异构烷烃的生成,C5+烃中异构烷烃的选择性高达92%,不期望产物芳烃的选择性可控制在6%以下。然而,两组分活性以颗粒方式混合或双床层方式装填时,烃类各产物的选择性变化不大,但颗粒混合时更有利于烃的生成。另一方面,粉末混合的复合催化剂上,芳烃选择性显著增加,即距离越近,越有利于芳烃生成,这与下文金属氧化物与分子筛串联催化剂上CO2加氢制芳烃变化趋势一致。而且,该核壳催化剂也表现出优异的稳定性,反应100 h,CO2的转化率及C5+烃的选择性均可维持在20%及50%左右,同时汽油烃中C5+异构烷烃及芳烃的选择性一直保持在90%以上及6%以下。因此,对于CO2加氢制异构烷烃及汽油反应来说,无论哪种串联催化剂,两活性组分间均不能太过接近,即两活性组分间需要保持一定的距离。
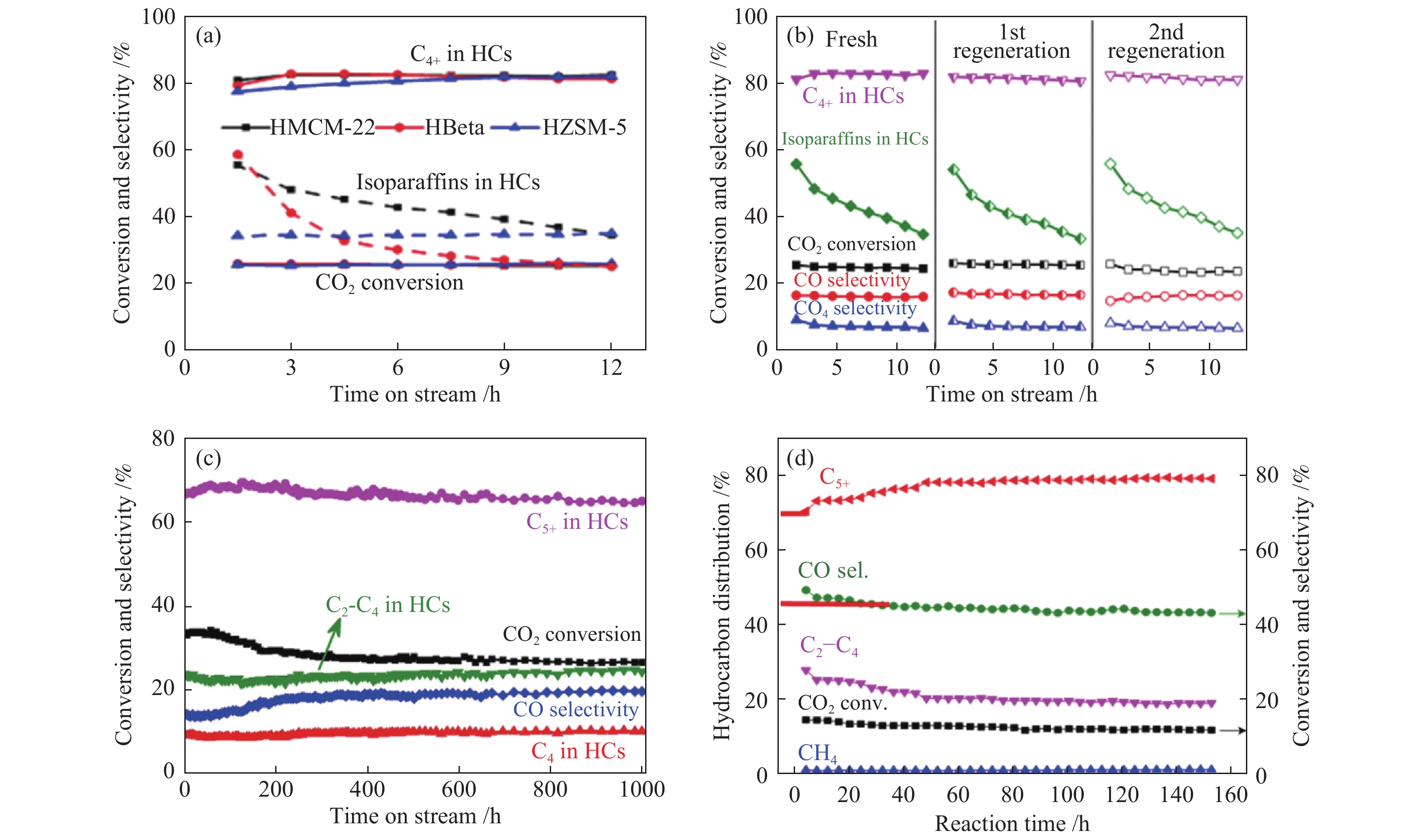
与CO2加氢制低碳烯烃类似,分子筛的结构及酸性同样对异构烷烃及汽油烃的选择性产生一定影响。Wei等[79]研究了Na-Fe3O4与不同孔道结构(HY、HBEA、HMOR、HZSM-23、 HMCM-22或HZSM-5)及硅铝比(HZSM-5(27)、HZSM-5(160)或HZSM-5(300))分子筛复合催化剂上CO2加氢制汽油烃的反应性能,在考察范围内,分子筛的孔道结构及酸性对CO2的转化率及CO的选择性均影响不大,但分子筛的孔道结构对烃产物分布影响显著,三种具有十元环孔道结构的分子筛(HZSM-5,HMCM-22,HZSM-23)最有利于C5–11的生成,C5–11的选择性依次为:HZSM-5(三维孔道)>HMCM-22(二维孔道)>HZSM-23(一维孔道)。分子筛的孔道结构一定,改变其硅铝比时,酸性适中的HZSM-5(160)分子筛最有利于C5-C11的生成。随后,Wei等[72]又报道了Na-Fe3O4与分子筛以双床层方式复合的串联催化剂上CO2加氢制异构烷烃性能。结果表明,Na-Fe3O4/HMCM-22及Na-Fe3O4/Hbeta复合催化剂上烃产物中异构烷烃的选择性明显高于Na-Fe3O4/HZSM-5,而Na-Fe3O4/HMCM-22催化剂上CO2转化率及C4+烃中异构烷烃的选择性分别为26%及74%(表2),这主要归因于三种分子筛不同的孔道结构及酸性。尽管HMCM-22(28)、Hbeta(24)及HZSM-5(25)三种分子筛的硅铝比相似,但其酸密度及酸强度明显不同;HZSM-5分子筛三维十元环(5.3 Å×5.6 Å,5.1 Å×5.5 Å)的孔道结构非常有利于烯烃芳构化生成芳烃,当二维层状十元环的HMCM-22分子筛及三维交叉十二元环孔道的Hbeta分子筛有利于烯烃聚合及异构化反应生成异构烷烃。
此外,对于核壳催化剂上CO2加氢制异构烷烃及汽油烃来说,分子筛的孔道结构及酸性对异构烷烃及汽油烃的生成同样具有显著影响[46,47]。Wang等[46]发现,Fe-Zn-Zr@Hbeta(4∶1)核壳催化剂非常有利于异丁烷(i-C4)的生成,烃中i-C4的选择性高达58%,i-C5+的选择性仅20%左右;而Fe-Zn-Zr@HZSM-5(4∶1)核壳催化剂有利于i-C5+的生成,烃中i-C5+的选择性可达35%。作者认为,烃中不同的异构烷烃分布主要因为两分子筛不同的孔道结构导致甲醇转化的反应机制不同,在反应温度下,Hbeta分子筛上甲醇转化易生成异丁烯,而HZSM-5分子筛上主要生成乙烯及丙烯。而且,Wang等[47]发现,HZSM-5分子筛的硅铝比及壳层厚度对串联催化剂的性能也具有显著影响。研究表明,CO2的转化率随HZSM-5分子筛的硅铝比变化不大,而适中的分子筛酸性有利于C5+烃中异构烷烃的生成。通过改变氧化物与分子筛的比例调节分子筛壳层的厚度,当HZSM-5的SiO2/Al2O3=50,需要调变分子筛的壳层厚度Fe-Zn-Zr/HZSM-5 = 6∶1−4∶1才能有利于C5+烃中异构烷烃的生成,而增加或降低分子筛壳层厚度均会降低C5+异构烷烃的选择性。分子筛壳层厚度一方面影响催化剂的酸性;另一方面影响中间物种与分子筛酸中心接触进行二次反应的程度,从而影响烃中各产物的分布。更重要的是,分子筛的酸性与壳层厚度间需要良好的匹配,若增加HZSM-5的酸量,必要适当调节其壳层厚度,才能有效促进C5+烃中异构烷烃的生成。
与CO2加氢制低碳烯烃相似,如上所述,CO2加氢转化制异构烷烃及汽油烃主要采用两类串联催化剂:一是,碱金属改性的Fe/zeolite串联催化剂,在此类催化剂上CO2加氢遵循FTS反应路径,CO2的转化率相对较高,副产物CO较低,但烃类产物中CH4及芳烃的选择性较高,因而降低烃中CH4及芳烃的选择性对提高汽油烃的收率及品质至关重要;二是OX/zeolite串联催化剂,与FTS反应路径的串联催化剂相比,甲醇反应路径的烃产物中CH4的选择性可控制到1%左右,同时芳烃的选择性被显著抑制,烃中总异构烷烃及汽油烃中异构烷烃的选择性均相对较高,但CO2的转化率较低,且副产物CO的选择性较高(>40%)。因此,抑制此类催化剂上副产物CO的生成,提高总烃的选择性极其重要。
对于CO2加氢制备异构烷烃反应来说,增加原料气的H2/CO2比例,有利于提高CO2加氢还原生成CO以及CO经FTS反应路径生成烃,从而提高CO2的转化[72]。而且,随着H2/CO2比例的增加,CO的选择性明显降低,同时C4+烃及异构烷烃的选择性也略微降低。Fe-Zn-Zr-T-24h@HZSM-5核壳催化剂上CO2加氢时,反应温度由320 ℃升高至370 ℃时,CO2的转化率及副产物CO的选择性均逐渐增加,而汽油烃中C5+异构烷烃的选择性出现先增加后变化不大的趋势,综合考虑,340 ℃反应温度对汽油烃中异构烷烃的生成较为有利[26]。此外,In2O3/HZSM-5复合催化剂上CO2加氢制汽油时,增加反应空速不仅提高了C5+烃的选择性,而且副产物CO的选择性也逐渐降低[80]。
除改变反应条件降低副产物CO的选择性外,Wang等[26]采用TPABr水溶液对Fe-Zn-Zr氧化物进行水热处理,希望通过改变氧化物自身的物化性质有效提高其合成甲醇的目的。结果表明,TPABr水热处理后的氧化物(Fe-Zn-Zr-T)对逆水煤气反应具有明显的抑制作用,甲醇的选择性显著提高,且与分子筛复合制备的核壳催化剂上烃的选择性也进一步提高。这些主要归因于水热处理后获得的Fe-Zn-Zr-T氧化物表面出现明显的Zn富集及Br残留,同时氧空穴含量增加,这些表面性质不仅促使氧化物表面吸附的H2/CO2比例增加,而且增强了HCOO*物种的吸附强度及CH3O*物种的脱附速率,从而促使CO2加氢向甲醇合成的方向进行,副产物CO被显著抑制。
另外,Gao等[80]进一步考察了原料气中CO浓度对In2O3/HZSM-5催化剂上CO2加氢性能的影响,随CO2+H2气氛中CO浓度的增加,CO2的转化率及C5+烃的收率均明显提高,DFT理论计算与实验结果共同证明,与H2还原气氛相比,CO气氛更有利于在In2O3(110)表面形成氧空穴,从而进一步提高CO2加氢的活性。然而,当原料气中CO/(CO2+CO) = 100%时,In2O3被还原成金属相,此时不能检测出C5+烃的生成。
积炭失活通常是分子筛上进行酸催化反应失活的主要原因。Wei等[72]发现,Na-Fe3O4/HMCM-22及Na-Fe3O4/Hbeta串联催化剂上进行CO2加氢制异构烷烃时,异构烷烃的初活性显著高于Na-Fe3O4/HZSM-5催化剂,然而,Na-Fe3O4/HMCM-22及Na-Fe3O4/Hbeta两催化剂在反应12 h时异构烷烃的选择性均明显降低,相比之下,Na-Fe3O4/HZSM-5催化剂稳定性较好(图9(a))。作者通过XRD、N2物理吸附表征发现分子筛的积炭物种主要存在微孔内,从而堵塞反应物与酸中心接触而逐渐失活。TPO及 TG表明,HMCM-22及Hbeta两分子筛反应12 h后的积炭量分别为14%及12%,明显高于HZSM-5分子筛3%的积炭量。分子筛酸中心上积炭的形成过程是一个复杂的择型催化反应,分子筛的孔结构起着非常重要的作用。也就是说,HMCM-22、Hbeta及HZSM-5三种分子筛上不同的积炭速率与它们拥有不同的孔结构密切相关,HMCM-22及Hbeta分子筛分别拥有超笼及较大孔穴,能够提供更大空间使碳链增长形成较大的烃类分子,进一步生成积炭导致分子筛失活。然而,HZSM-5分子筛相互交叉的孔道结构可抑制大分子积炭中间体形成,因此,具有较低的积炭速率。此外,作者对Na-Fe3O4/HMCM-22串联催化剂进行两次再生实验发现,再生后异构烷烃的选择性及CO2转化率均可恢复至初始水平,但仍具有较快的失活速率(图9(b))。
Wei等[79]发现,Na-Fe3O4/HZSM-5(160)串联催化剂在CO2加氢制汽油燃料时表现出极高的稳定性(图9(c))。在320 ℃,3 MPa,4000 mL/(gcat·h)条件下连续运行1000 h,C5+烃保持(67±2)%选择性,CO2转化率仅在反应最初的300 h下降了6%,这主要归因于反应初期活性Fe发生迁移导致其比表面积下降,随后Fe物种保持稳定导致CO2转化率变化不大。而对于HZSM-5分子筛来说,反应1000 h积炭量为3.7%,此串联催化剂极高的催化活性及稳定性赋予其非常好的工业应用潜力。In2O3/HZSM-5[80]串联催化剂在CO2加氢制汽油燃料反应中也展现出非常好的稳定性(图9(d))。在反应最初40 h时,CO2转化率及CO选择性有所降低,C5+烃选择性增加,这主要归因于反应初期In2O3晶粒尺寸的增大;随着反应时间延长至150 h,CO2转化率及C5+烃的选择性分别保持在12%及80%。除此之外,Wang等[47]发现,Fe-Zn-Zr@HZSM-5核壳催化剂在CO2加氢制高辛烷值汽油反应中也具有良好的催化稳定性。因此,Fe基或氧化物与HZSM-5复合形成的串联催化剂均具有很好的工业应用前景。
(a): Fresh Na-Fe3O4/zeolite[72]; (b): Regenerated Na-Fe3O4/HMCM-22[72]; (c): Na-Fe3O4/HZSM-5[79]; (d): In2O3/HZSM-5[80] reproduced with permission from ref. [72, 79, 80], Copyright (2018) American Chemical Society, Copyright (2017) Springer Nature
芳烃作为非常重要的平台化合物,可以用来制备聚苯乙烯、酚醛树脂、尼龙等各种聚合物。将CO2加氢催化转化生成芳烃是值得发展的绿色芳烃制备技术,与CO2加氢制备异构烷烃及汽油反应类似,同样属于多步串联催化过程,采用的催化剂也主要分为两类,如表3所示:Fe/zeolite串联催化剂;OX/zeolite串联催化剂。而且,不同的串联催化剂上CO2加氢制芳烃分别遵循CO中间体的FTS反应路径或甲醇及甲醇前驱物为中间体的反应路径。然而,与CO2加氢制低碳烯烃及异构烷烃不同的是,CO2加氢芳烃在分子筛酸中心上转化的路径更长,实施催化反应需要更长的接触时间,且更容易发生副反应[2]。
如表3 所示,对于Fe/zeolite串联催化剂来说,Fe基催化剂通常需要进行碱金属改性,而且碱金属离子的含量对芳烃选择性影响较大。Cui等[81]考察了ZnFeOx-Na及HZSM-5分子筛串联催化剂上CO2加氢制芳烃的性能。研究发现,ZnFeOx-Na氧化物上CO2加氢时,随着Na离子含量的增加,CO2的转化率及烯烃/烷烃(O/P)摩尔比率均先增加后降低,Na离子含量为2.68%及4.25%时,CO2的转化率均达38.5%,且Na离子含量为4.25%时,O/P比率高达7.7。Fe3O4及Fe5C2是反应过程中主要的活性相,CO2首先在Fe3O4相上还原生成CO,然后CO在Fe5C2相上经FTS生成低碳烯烃,仅Fe5C2是FTS的活性相,对CO2还原并不活泼,而且Fe5C2相的形成与Na离子含量密切相关(图10(a))。因此,作者推断,Fe3O4及Fe5C2两相间恰当的比例是ZnFeOx-2.68Na及ZnFeOx-4.25Na氧化物上CO2转化率及O/P比率高的原因。这与上文Xu等[71]报道的Fe-Cu-Na/zeolite复合催化剂上CO2加氢制异构烷烃的反应趋势相似,适中的Na离子含量更有利于低碳烯烃的生成。
下载:
导出CSV
| Catalyst | t /℃ | p /MPa |   | saromatics /% |
| ZnFeOx-4.25Na/S-HZSM-5 (Granule-mixing)[81] | 320 | 3 | 41.2 | 75.6 |
| Na/Fe-HZSM-5 (Granule-mixing)[82] | 320 | 1 | 29.4 | 54.3 |
| FeK1.5/HSG-HZSM-5 (Dual-bed)[83] | 340 | 2 | 35.0 | 68.0 |
| 6.25Cu-Fe2O3/HZSM-5 (Granule-mixing)[84] | 320 | 3 | 57.3 | 56.6 |
| ZnZrO/ZSM-5 (Powder-mixing)[40] | 320 | 4 | 14.0 | 73.0 |
| ae-ZnO-ZrO2/Z5 (Powder-mixing)[85] | 340 | 4 | 16.0 | 76.0 |
| ZnO/ZrO2-ZSM-5 (Powder-mixing)[86] | 340 | 3 | 9.0 | 70.0 |
| ZnAlOx & HZSM-5 (Powder-mixing)[87] | 320 | 3 | 9.1 | 73.9 |
| Cr2O3/HZSM-5 (Powder-mixing)[27] | 350 | 3 | 34.5 | 76.0 |
| ZnCrOx-ZnZSM-5 (Powder-mixing)[88] | 320 | 5 | 19.9 | 81.1* |
| * Aromatics selectivity in C5+ hydrocarbons, the others are the aromatics selectivity in all hydrocarbons | ||||

(a): ZnFeOx-4.25Na/S-HZSM-5[81]; (b): 6.25Cu-Fe2O3/HZSM-5[84]; (c); ZnZrO/HZSM-5[40] reproduced with permission from ref. [81, 84, 40], Copyright (2019 & 2020) American Chemical Society, Copyright (2019) Elsevier
除Na离子外,K离子改性的Fe与分子筛复合的串联催化剂也被用于CO2加氢制芳烃反应[83]。与Na离子相似,K离子同样需要一个恰当的含量,过高或过低K含量的FeK1.5/HSG与HZSM-5分子筛复合时均不利于芳烃生成。除碱金属Na及K离子改性的Fe基催化剂用于CO2加氢制芳烃外,Song等[84]发现,Cu改性的Fe基催化剂与HZSM-5分子筛复合制备的6.25Cu-Fe2O3/HZSM-5-c催化剂上CO2加氢也可高选择性合成芳烃。在该复合催化剂上,CO2加氢同样遵循RWGS及FTS路径(图10(b)),Fe2O3首先经CO还原生成Fe3O4及Fe2C5,CO2在两相上经RWGS及FTS连续反应生成低碳烯烃,然后在分子筛上进一步聚合、芳构化生成芳烃。恰当的Cu含量不仅不影响CO的吸附,而且有利于CO的活化及进一步向烯烃中间体的转化,从而导致高的CO2转化率及低的CO选择性。上文作者提到,Fe2C5相的生成与碱金属离子的含量密切相关,然而,对于6.25Cu-Fe2O3催化剂来说,并不存在碱金属离子,Fe2C5相的生成主要归于催化剂在CO气氛下的还原处理。因此,对于Fe/zeolite串联催化剂上CO2加氢反应来说,碱金属离子可能并非必须存在,CO转化的Fe2C5活性相可通过催化剂在CO气氛下预处理获得。
如表3所示,不同的金属氧化物与分子筛复合形成的串联催化剂上CO2加氢制芳烃时同样可高选择性的合成芳烃,在此类串联催化剂上,CO2加氢通过甲醇或甲醇前驱物种(CHxO)在分子筛酸中心上进行连续转化生成芳烃。Li等[40]报道的ZnZrO固溶体与HZSM-5分子筛串联催化剂上,CO2的单程转化率及芳烃选择性分别达14%及73%。通过原位红外表征分别对ZnZrO固溶体及ZnZrO/HZSM-5复合催化剂上CO2加氢时生成的表面物种进行研究,认为CO2加氢在ZnZrO固溶体上主要生成甲醇的前驱物种CHxO(CH3O*、CHO*及气相CH3OH),而且,通过NH2-SBA对CHxO物种捕获进一步证明了CHxO物种从ZnZrO表面通过分子筛的孔道在其酸中心上进行一系列的酸催化反应生成芳烃(图10(c))。Zhou等[85]通过溶胶凝胶及超临界干躁相结合的方法制备的ZnO-ZrO2氧化物拥有较大的比表面积及丰富的氧空穴,因此与HZSM-5分子筛复合制备的串联催化剂上CO2加氢时,在340 ℃,4 MPa条件下,CO2的转化率及芳烃的选择性分别达16%及76%,副产物CO的选择性低至34%,芳烃的收率高达0.24 g/(goxide·h)。在此串联催化剂上,CO2主要在ZrO2上进行吸附活化,ZnO主要起解离H2的作用,CO2加氢通过甲醇中间体进行转化生成芳烃,甲醇的形成速率由氧化物上总的氧空穴量决定。此外,ZnO/ZrO2[86]、ZnAlOx[87]、Cr2O3[27]及ZnCrOx[88]等氧化物与HZSM-5分子筛复合催化剂上CO2加氢制芳烃时,芳烃的选择性也可达70%以上,此类串联催化剂上芳烃的选择性通常高于Fe基与分子筛串联催化剂上芳烃的选择性。
由表3 可知,Fe/HZSM-5或OX/HZSM-5串联催化剂上CO2加氢制芳烃时两组分间的组装方式截然不同,这可能与两类串联催化剂上CO2加氢的反应路径有关。Cui等[81]报道了ZnFeOx-4.25Na/S-HZSM-5串联催化剂上两类活性组分组装方式对催化性能的影响(图11(a))。ZnFeOx-Na及HZSM-5从双床层构型到颗粒方式混合时,两组分间的距离逐渐接近,串联催化剂上CO2的转化率及芳烃的选择性均明显增加;随着两组分间距离的进一步接近,以粉末方式混合时,CO2的转化率及芳烃的选择性反而降低,说明ZnFeOx-Na及HZSM-5串联催化剂上CO2加氢制芳烃时,并非是距离越短越好。两活性组分间的距离较短,ZnFeOx-Na上的Na离子会迁移到分子筛上,毒化分子筛的酸中心,降低分子筛中强酸中心的数量,导致分子筛快速失活及芳烃选择性降低。当ZnFeOx-Na及HZSM-5通过玛瑙研钵进行研磨混合,两组分间的距离更加接近时,CO2的转化率及芳烃的选择性进一步降低,充分证明ZnFeOx-Na/HZSM-5串联催化上CO2加氢制芳烃时,两组分间的距离不能太过接近。而且,在6.25Cu-Fe2O3/HZSM-5-c 串联催化剂上CO2加氢制芳烃时显示出同样的规律(图11(b))[84]。6.25Cu-Fe2O3及HZSM-5-c两组分以颗粒方式混合时,CO2的转化率、芳烃及C5+烃的选择性均最佳,分别为57.3%、56.61%及80.89%。通过Cu的辅助作用,副产物CO的选择性降低至3.51%。当6.25Cu-Fe2O3及HZSM-5-c以粉末方式混合时,两活性组分间的距离非常接近,CO2的转化率及芳烃选择性均明显降低,同时不期望的CH4选择性急剧增加,其主要认为两活性组分距离太短时,HZSM-5分子筛的酸中心毒化6.25Cu-Fe2O3表面的碱中心,降低氧化物的碱性。因此,两组分间的距离过长或过短,即石英砂稀释、双床层或粉末混合时,均不利于CO2的转化及芳烃的生成。另外,Wang等[83]也发现,FeK1.5/HSG与HZSM-5以双床层方式结合时,串联催化剂的催化性能明显优于两组分均一混合时的催化性能(图11(c))。在340 ℃,2.0 MPa,26000 mL/(gcat·h)条件下,双床层催化剂上CO2的单程转化率达35%,所有含碳产物中(包括CO)及烃产物中芳烃的选择性分别为41%及68%。在该串联催化剂上,CO2加氢首先在FeK1.5/HSG组分上生成烯烃中间体,然后烯烃中间体扩散到分子筛孔道中经聚合、环化及脱氢等过程生成芳烃,烯烃是芳烃直接的前驱物种,两组分间紧密的接触不必要(图11 (d))。
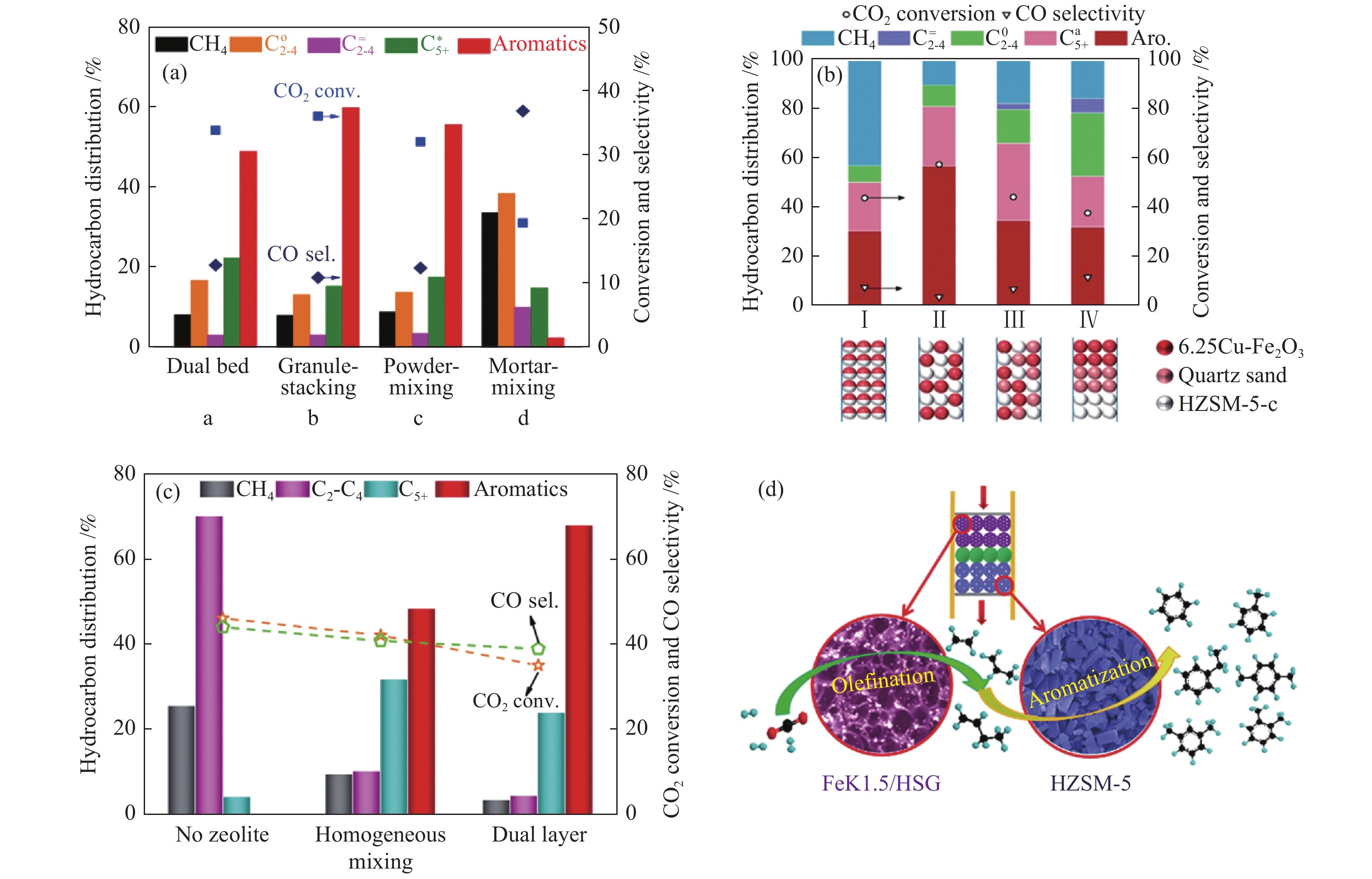
(a): ZnFeOx-4.25Na/S-HZSM-5[81]; (b): 6.25Cu-Fe2O3/HZSM-5-c[84]; (c), (d): FeK1.5/HSG-HZSM-5(50)[83] reproduced with permission from ref. [81, 84, 83], Copyright (2019 & 2020 & 2019) American Chemical Society
金属氧化物与分子筛串联催化剂上CO2加氢制芳烃时,两活性组分间的接近度同样是影响芳烃生成的关键因素。然而,与CO中间体的FTS反应路径不同的是,氧化物及HZSM-5分子筛串联催化剂上CO2加氢制芳烃时,两活性组分均以粉末方式结合时展现出优异的芳烃选择性[27,40,85-88]。Li等[40]研究了ZnZrO/HZSM-5串联催化剂上CO2加氢制芳烃的催化性能,在320 ℃,3 MPa,1200 mL/(gcat·h)反应条件下,两活性组分以粉末方式混合时,CO2的单程转化率及烃类产物中芳烃的选择性分别达14%及73%,显著高于颗粒混合及双床层混合的催化剂(图12(a))。此外,日本Noritatsu Tsubaki课题组的Wang等[27]及中国科学院大连化学物理研究所刘中民课题组的Ni等[87]也发现,Cr2O3/H-ZSM-5粉末混合催化剂及ZnAlOx&HZSM-5粉末混合催化剂上CO2加氢制芳烃时,CO2的转化率及烃中芳烃的选择性均明显高于颗粒混合及双床层催化剂((图12(b)、(c))。此外,ZnCrOx-zeolite粉末混合催化剂上CO2加氢也具有良好的芳烃选择性[88]。因此,对于金属氧化物与H-ZSM-5串联催化剂来说,两组分间需要非常短的距离来促进芳烃的生成。这与前文金属氧化物与分子筛串联催化剂上CO2加氢制低碳烯烃的结论相一致,均需要两活性组分间的充分接触才能有利于目标产物的生成。究其根本原因,CO2加氢制低碳烯烃与芳烃反应均通过甲醇中间体进行转化,而甲醇转化的驱动力与氧化物及分子筛两组分间的接近度密切相关。
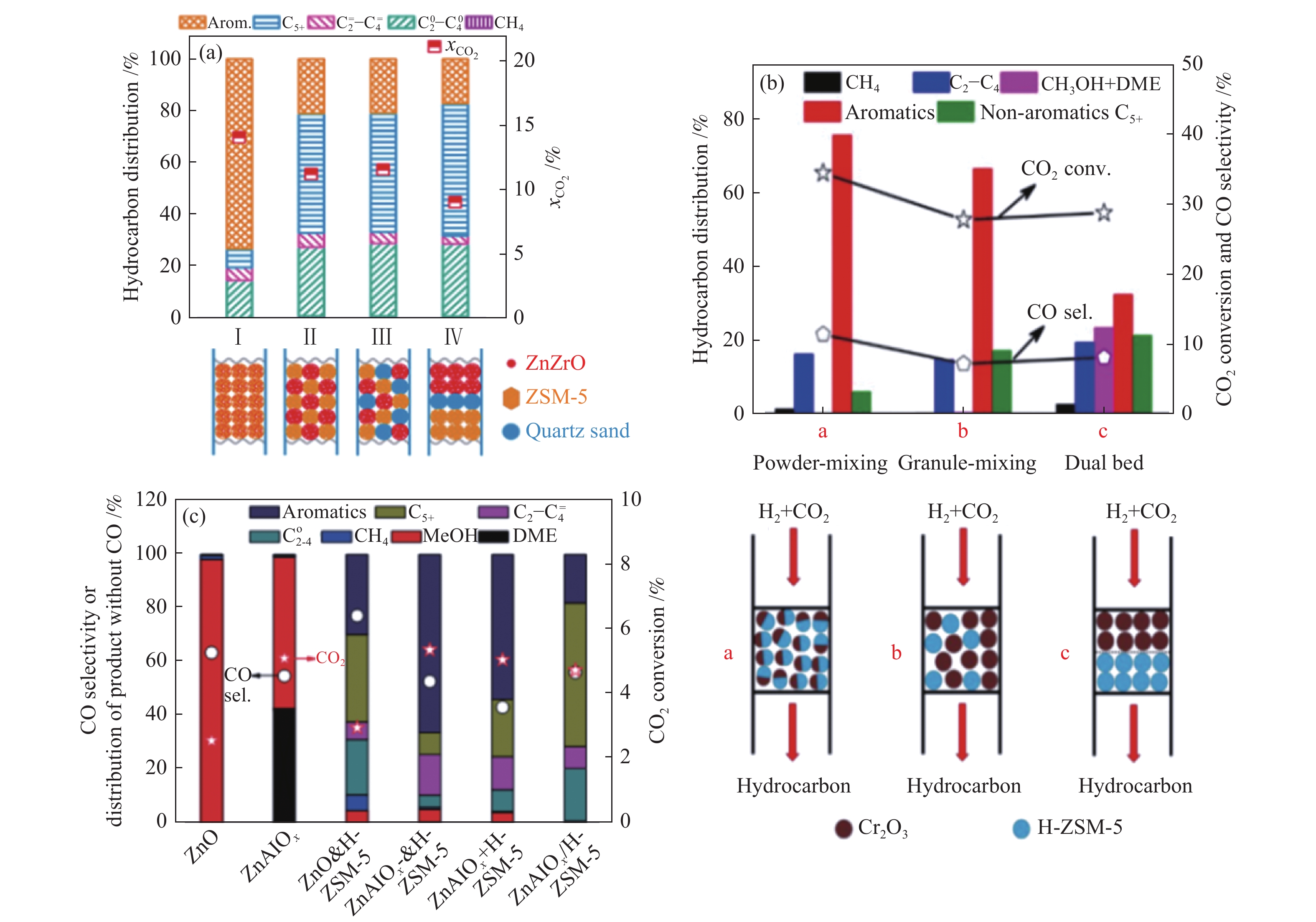
(a): ZnZrO/HZSM-5[40]; (b): Cr2O3/HZSM-5[27]; (c): ZnAlOx&HZSM-5[87] reproduced with permission from ref. [40, 27, 87], Copyright (2019) Elsevier, Copyright (2019) American Chemical Society
因此,在碱金属离子或Cu改性的Fe基与HZSM-5分子筛串联催化剂上,CO2加氢制芳烃均遵循CO中间体的反应路径,两类活性组分间均不能太过接近,以双床层或颗粒混合方式复合时,串联催化剂的性能最佳;两组分以非常近的距离,即粉末方式混合时,烃产物中不期望的CH4选择性急剧增加,不利于目标产物的生成,两组分间需要保留恰当的距离。然而,对于ZnZrO、ZnAlOx、Cr2O3及ZnCrOx等氧化物与HZSM-5分子筛串联催化剂来说,CO2加氢制芳烃均遵循甲醇或CHxO中间体的反应路径,金属氧化物与分子筛两组分间的距离越接近,越有利于生成芳烃。总之,对于Fe基催化剂来说,一是其具有较强的加氢能力;二是在Fe基催化剂上CO2加氢直接生成低碳烯烃,分子筛的酸中心仅起低碳烯烃聚合、环化及芳构化的作用,这些反应并不需要两活性组分间的亲密接触。然而,金属氧化物的活性中心与分子筛界面间可能存在一定的协同作用,在甲醇或CHxO中间体的转化过程中不单单是分子筛的酸中心起作用[34],两类活性组分间的紧密接触非常重要。
串联催化剂上CO2加氢制芳烃无论遵循哪一条反应路径,反应中间体在分子筛的酸中心上均进行一系列的酸催化反应,因此,分子筛的结构及酸性同样对芳烃产物的生成及分布影响显著。Li等[40]发现,ZnZrO/HZSM-5串联催化剂上芳烃的选择性与HZSM-5分子筛的酸性密切相关,随着HZSM-5硅铝比的增加,芳烃的选择性先增加后降低,Si/Al = 100时,芳烃的选择性最高,过高或过低的硅铝比均不利于芳烃的生成。而且,分子筛的孔道结构对芳烃的生成也具有明显影响。Wang等[83]研究了FeK1.5/HSG与不同结构分子筛(SAPO-34、HY、Hβ、HMCM-22或HZSM-5)串联催化剂上CO2加氢制芳烃的性能(图13(a))。研究表明,FeK1.5/HSG与SAPO-34分子筛复合时,由于SAPO-34分子筛最小的孔道尺寸(3.8 Å×3.8 Å),导致烃产物中芳烃的选择性最低。HY及Hbeta分子筛均具有十二元环的孔道结构(7.4 Å× 7.4 Å,6.6 Å×6.7 Å),但Hbeta具有额外较小的十二元环孔道(5.6 Å×5.6 Å),与芳烃的动力学直径接近,具有更好的孔道择形性,所以FeK1.5/HSG|Hbeta串联催化剂上芳烃的选择性较FeK1.5/HSG|HY更高。此外,对于同是十元环孔道的HMCM-22(二维孔道,4.0 Å×5.5 Å,4.1 Å×5.1 Å)及HZSM-5(三维孔道,5.1 Å×5.5 Å,5.3 Å × 5.6 Å)分子筛来说,HMCM-22分子筛由于较小的孔道尺寸,导致串联催化剂上芳烃的选择性略低于HZSM-5分子筛。因此,作者认为芳烃的选择性主要与分子筛的孔道尺寸密切相关,而分子筛二维或三维的孔道结构及十元或十二元的孔道环数对芳烃的选择性来说并不是决定性因素,最佳的孔道尺寸是5–6 Å。另外,通过对比不同硅铝比的HZSM-5分子筛,发现SiO2/Al2O3 = 50时,芳烃的选择性最高,即分子筛的孔道结构相同时,分子筛酸性成为影响芳烃选择性的主要因素。此外,不同的金属氧化物与HZSM-5分子筛复合时,芳烃性能最佳时HZSM-5分子筛的酸性并不相同,这均归因于两活性组分间的不同耦合程度。
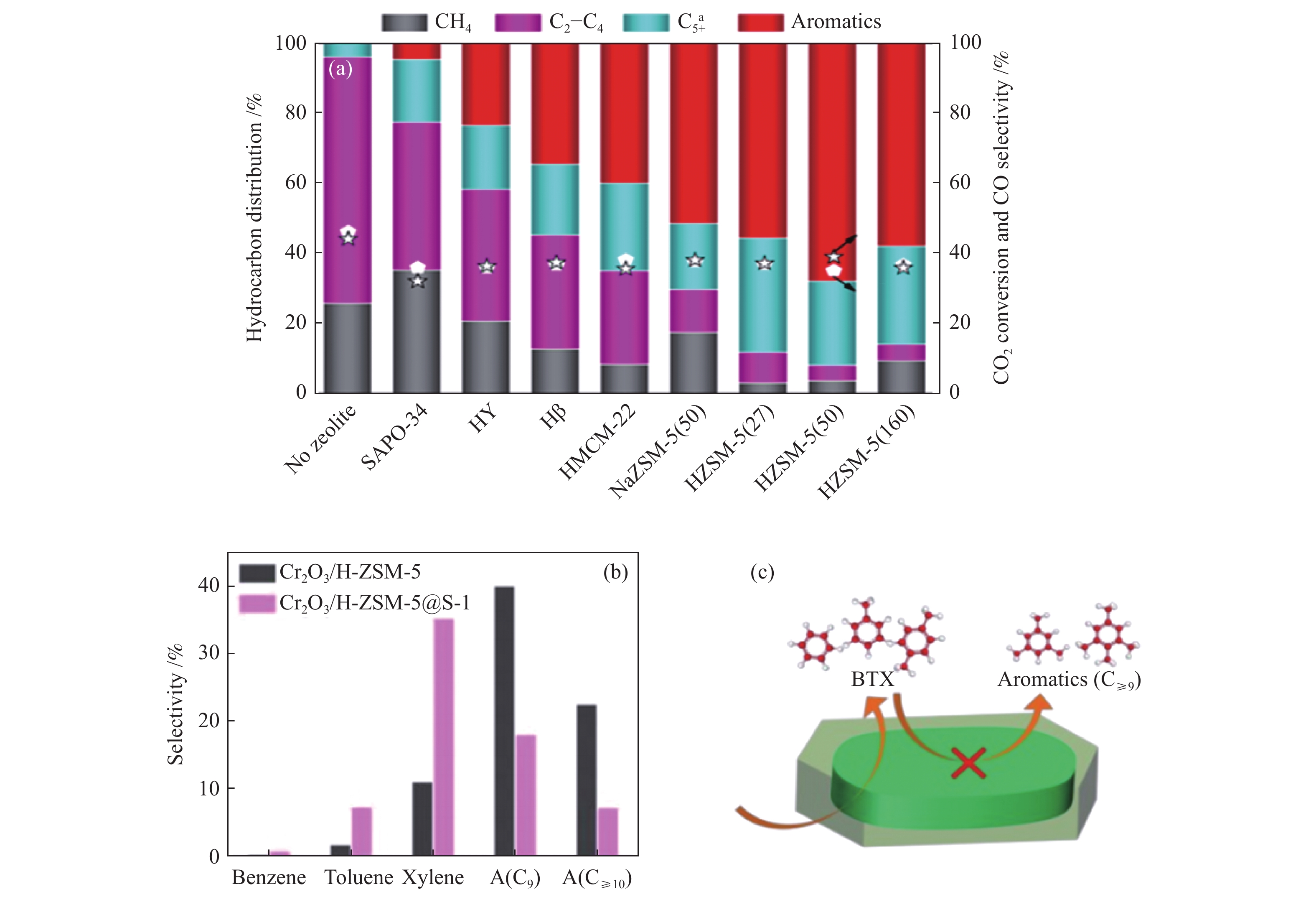
除了改变HZSM-5分子筛的硅铝比来调变总芳烃选择性外,对分子筛外表面的酸中心进行调变也可进一步提高芳烃中轻质芳烃(BTX)的选择性。Wang等[27]通过在HZSM-5表面包覆纯硅S-1分子筛覆盖HZSM-5外表面的酸中心,抑制不期望的烷基化及异构化反应,从而提高芳烃中BTX的选择性。如图13(b)、(c)所示,Cr2O3与核壳结构的H-ZSM-5@S-1分子筛复合时,BTX的选择性显著提高,同时C≥9芳烃的选择性明显降低,BTX及对二甲苯的选择性分别由13.2%及7.6%提高至43.6%及25.3%。此外,将脱氢组分Zn组分引入到HZSM-5分子筛中,同时在Zn-ZSM-5分子筛外包覆上无定型的SiO2壳,覆盖分子筛外表面的酸中心,可进一步提高芳烃中对二甲苯的选择性[27]。Cr2O3/Zn-ZSM-5@SiO2串联催化剂上烃中对二甲苯的选择性提高至38.7%,CO2转化率达22.1%。
与CO2加氢制备低碳烯烃、异构烷烃及汽油烃类似,CO2加氢制芳烃时,随反应温度的升高,CO2的转化率及副产物CO的选择性均呈现逐渐增加趋势。反应温度较高时,低碳烯烃的氢转移及甲基化反应增强,从而导致低碳烷烃及C5+烃的选择性增加,芳烃的选择性明显降低[40]。随反应空速的增加,CO2的转化率明显下降,芳烃的选择性先增加后降低。而且,增加原料气的H2/CO2比例有利于CO2的转化及副产物CO的抑制,芳烃的选择性有所降低,其原因同样是较高的H2分压有利于低碳烯烃发生氢转移反应,而发生聚合、环化及芳构化反应的程度降低。
为了深入研究CO2加氢制芳烃的反应机制及关键影响因素,Li等[40]进一步对比了串联催化剂上CO2加氢制芳烃及甲醇转化制芳烃的性能差异。研究发现,CO2加氢时芳烃的选择性显著高于甲醇芳构化时芳烃的选择性,其主要原因为两者不同的反应气氛,CO2加氢过程中有大量H2O生成及CO2气氛。作者通过对比HZSM-5分子筛上乙烯及不同含量H2O共进料时芳烃的选择性发现,随着H2O含量的增加,芳烃选择性出现先增加后降低的趋势,说明恰当的H2O含量有利于烯烃进一步芳构化生成芳烃。这主要归因于H2O分子和乙烯分子在弱酸中心上存在竞争吸附,当H2O分子吸附于弱酸中心上时,将有利于乙烯分子在分子筛的强酸中心上进行吸附及芳构化反应生成芳烃。
与此同时,CO2加氢芳构化过程中COx(CO、CO2)的存在对芳烃的生成及副产物CO的抑制同样具有明显的促进作用。Li等[40]进一步对比了甲醇、乙烯与H2共进料时芳烃的选择性,发现反应气氛中H2的存在会抑制芳烃的生成,因而分子筛上烯烃脱氢时,氢物种的去除是烯烃芳构化的速控步骤。而CO2的存在可有效移除烯烃芳构化过程中脱除的氢,从而明显提高芳烃的生成。Wang等[27]发现,在金属氧化物及分子筛双功能催化剂上CO2加氢时,原料气中添加CO不仅能够有效提高芳烃的选择性,而且同时抑制了逆水煤气反应的进行,CO的这种作用归因于对化学平衡的拉动以及有利于氧空穴的循环产生。另外,通过对单纯HZSM-5分子筛及Cr2O3/HZSM-5串联催化剂分别在N2及CO2气氛下芳构化的进一步研究(图14(a))[89],发现N2气氛下甲醇在HZSM-5及Cr2O3/HZSM-5催化剂上转化时芳烃的选择性均较低,而CO2气氛下甲醇在Cr2O3/HZSM-5催化剂上芳烃的选择性与单纯的HZSM-5催化剂相比明显提高。作者阐明了串联催化过程合成芳烃的CO2-辅助作用示意图(图14(b))。串联催化剂上CO2加氢生成的低碳烯烃是芳烃合成的主要中间体,低碳烯烃的转化路径主要有两条:一是低碳烯烃直接加氢生成烷烃;二是脱氢、环化生成芳烃。然而,在CO2气氛下,吸附在Cr2O3上的CO2可以及时去除低碳烯烃脱氢环化过程中产生的氢物种,从而进一步促进烯烃转化生成芳烃,抑制烷烃的生成,同时Cr2O3的出现对CO2的吸附活化也具有促进作用。与之相似,王野课题组的Cheng等[36]在Zn-ZrO2/HZSM-5串联催化剂上CO加氢制芳烃时,芳烃选择性可高达80%,但甲醇在此催化剂上反应时,芳烃的选择性较低,而甲醇在CO辅助进料时能够明显提高芳烃的选择性,其主要归因于CO的自催化作用。由此可见,在串联催化剂上,反应体系内的COx(CO、CO2)对芳烃的生成可能均具有非常重要的促进作用。

图15展示了不同串联催化剂上CO2加氢制芳烃的稳定性测试。Li等[40]发现,ZnZrO/ZSM-5串联催化剂在CO2加氢制芳烃反应中连续运行100 h,CO2转化率略有降低,而芳烃的选择性变化不大,说明此串联催化剂具有良好的稳定性(图15(a))。与之相似, ZnAlOx&H-ZSM-5[87]及ZnO/ZrO2-Z5-300[86]串联催化剂在CO2加氢制芳烃反应中同样表现出很好的稳定性,随着反应的进行,CO2转化率均变化不大,芳烃的选择性出现小幅度的降低(图15(b)、(c))。与氧化物/分子筛串联催化剂不同,碱金属改性的Fe基催化剂在CO2加氢反应中通常具有非常高的CO2活性。Cui等[81]发现,ZnFeOx-4.25Na/S-HZSM-5串联催化剂在CO2加氢制芳烃反应中连续反应100 h,CO2转化率始终保持35%以上,同时芳烃选择性也是小幅度降低(图15(d))。作者发现,串联催化剂的稳定性与HZSM-5的晶粒尺寸及孔结构密切相关。ZnFeOx-4.25Na与大晶粒的C-HZSM-5复合时,ZnFeOx-4.25Na/C-HZSM-5串联催化剂连续反应约72 h时,尽管CO2转化率保持在35%左右,但芳烃选择性明显降低,由最初的50%降至10%;通过NaOH溶液对C-HZSM-5分子筛进行碱处理引入介孔孔道制备出C-HZSM-5-a时,ZnFeOx-4.25Na/C-HZSM-5-a串联催化剂的稳定性明显提高,尽管芳烃的初始选择性略低(约45%),但连续反应72 h时,芳烃选择性仍可保持在30%以上。因此,与CO2加氢制低碳烯烃及汽油烃类似,串联催化剂上CO2加氢制芳烃时,CO2转化率的变化主要与Fe基或氧化物活性物种的变化相关,而芳烃选择性的变化主要与分子筛的积炭速率密切相关,降低HZSM-5的晶粒尺寸或引入多级孔道可显著提高串联催化剂的稳定性。
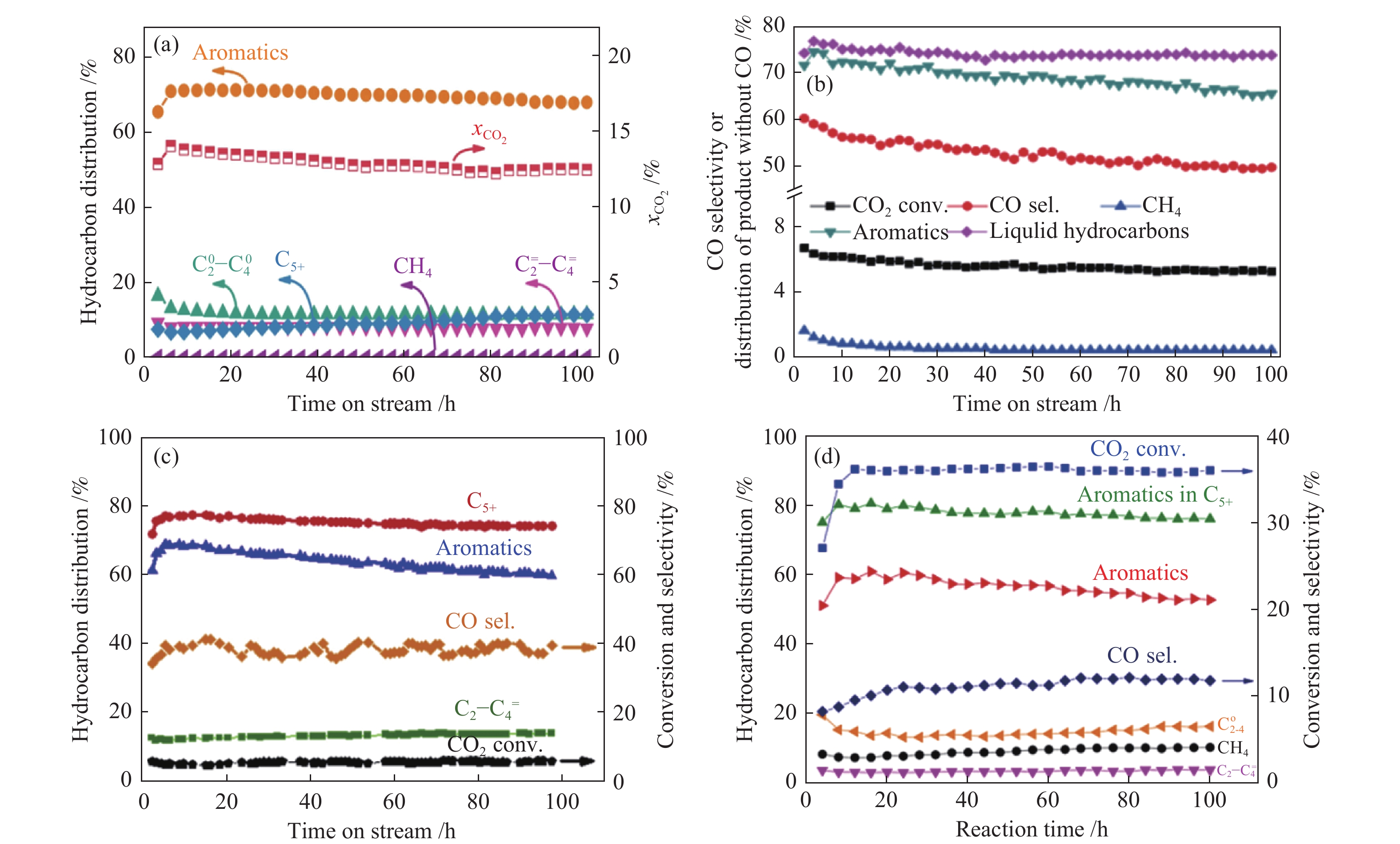
(a) ZnZrO/ZSM-5[40]; (b) ZnAlOx&H-ZSM-5[87]; (c) ZnO/ZrO2-ZSM5[86]; (d) ZnFeOx-4.25Na/S-HZSM-5[81] reproduced with permission from ref. [40, 87, 86, 81], Copyright (2019 & 2019) Elsevier, Copyright (2019) American Chemical Society
将温室气体CO2通过催化加氢的方法转化制备高附加值化学品或液体燃料,是实现碳资源循环利用、缓解能源危机以及应对双碳目标的有效途径之一。近年来,利用可再生能源(生物质、太阳能或风能等)发电和水电解产生的氢气,把CO2催化转化为高附加值化学品和燃料在环保及替代化石资源利用等方面意义重大。相对于C1产品来说,CO2加氢制备高碳烃的过程更加复杂及困难,而串联催化策略的出现将多步串联反应耦合在一起,通过活性组分间的高效协同拉动反应的进行,从而实现CO2加氢到C2+烃的高选择性合成。在CO2加氢制备C2+烃的不同串联催化过程中,Fe基催化剂或金属氧化物与分子筛组分间的良好匹配、两类活性组分间的组装方式、分子筛的孔道结构及酸性、以及反应条件与气氛均对烃类产物的分布影响显著(如图16所示)。

CO2加氢制低碳烯烃反应主要采用金属氧化物与分子筛串联催化剂,氧化物表面具有的丰富氧空穴可有效的活化CO2分子,当与八元环结构的H-RUB-13或SAPO-34分子筛耦合时实现了CO2加氢到低碳烯烃的高选择性合成。两类活性组分间的组装方式与氧化物自身性质密切相关,具有较高加氢活性的氧化物与分子筛复合时,两活性组分间的距离不能太过接近,太短的距离会导致低碳烯烃进一步加氢生成烷烃,降低目标产物的选择性;而氧化物加氢活性较低时,两组分需要以最近的接近度来促进中间产物的转化,提高低碳烯烃
CO2加氢制异构烷烃或汽油主要采用碱金属改性的Fe/zeolite或OX/zeolite串联催化剂,在这两类串联催化剂上CO2加氢主要遵循CO中间体的 FTS反应路径或甲醇中间体反应路径。无论哪种串联催化剂,两类活性组分间的距离均不能太过接近,即两类活性组分间需要保持一定的距离。两组分以非常短的距离,即粉末方式结合时,烃产物中芳烃或不期望的CH4选择性急剧增加,从而导致异构烷烃或汽油烃的选择性显著降低。尽管目前已报道的CO2加氢制异构烷烃及汽油燃料的串联催化剂较少,但通常可用于CO2加氢制备低碳烯烃及芳烃的甲醇合成氧化物,只要能够实现其与分子筛间的良好匹配,均有希望达到较好的异构烷烃及汽油性能。通过改变甲醇合成氧化物表面HCOO*及CH3O*物种的吸附-脱附速率,可有效促进CO2加氢向甲醇合成方向转化,进一步抑制副产物CO的生成。串联催化剂的稳定性主要取决于分子筛的积炭速率,具有较低积炭速率的HZSM-5分子筛在CO2加氢制异构烷烃或汽油烃反应中均具有非常好的稳定性。
与CO2加氢制异构烷烃相似,Fe/zeolite串联催化剂上CO2加氢制芳烃时,两类活性组分间同样不能太过接近,太近的接近度会导致活性组分彼此间的离子迁移或酸碱性变化,从而降低芳烃选择性。OX/zeolite串联催化剂上CO2加氢制芳烃时,两类活性组分以粉末方式结合时(最接近的距离),最有利于目标产物的生成,即距离越短越好。芳烃的选择性与分子筛的孔道尺寸密切相关,分子筛二维或三维的孔道结构及十或十二元的孔道环数对芳烃选择性来说并不是决定性因素,最佳的孔道尺寸为5-6 Å。通过对HZSM-5分子筛外表面酸性的包覆处理,可有效提高轻质芳烃(BTX)及对二甲苯(PX)的选择性,且反应过程中H2O及COx(CO、CO2)的存在对芳烃生成均存在明显的促进作用。与CO2加氢制异构烷烃及汽油烃类似,串联催化剂在CO2加氢制芳烃反应中的稳定性与HZSM-5分子筛的积炭速率密切相关,降低HZSM-5的晶粒尺寸或引入多级孔道可显著提高串联催化剂的稳定性。
总之,无论CO2加氢制备低碳烯烃、异构烷烃、汽油抑或芳烃,串联催化体系的构建及活性组分间的良好匹配共同实现了目标产物的高选择性合成。然而,针对串联催化体系,不同的催化剂组成、元素配比、制备方法以及目标产物等均影响两类活性组分间的最佳结合方式,而活性组分间距离究竟如何影响串联催化剂表面中间物种的生成、演变及迁移规律;与此同时,CO2加氢反应过程中,CO2、CO及H2O同时存在,三者在反应过程中对中间物种的生成及反应机制的影响作用如何,这些均是值得深入研究及探讨的问题。因此,简化反应体系或开展更加直观的原位表征手段对CO2加氢过程中间产物的生成及演变规律进行详细而深入的研究是未来发展的主要趋势。
SHA F, HAN Z, TANG S, WANG J, LI C. Hydrogenation of carbon dioxide to methanol over non Cu-based heterogeneous catalysts[J]. ChemSusChem,2020,13:6160−6181.
成康, 张庆红, 康金灿, 王野. 二氧化碳直接制备高值化学品中的接力催化方法[J]. 中国科学:化学,2020,50(7):743−755. doi: 10.1360/SSC-2020-0053CHENG Kang, ZHANG Qing-hong, KANG Jin-can, WANG Ye. The tandem catalysis of CO2 conversion to high value–added chemicals[J]. Sci Sin Chim,2020,50(7):743−755. doi: 10.1360/SSC-2020-0053
BANDO K, SOGA K, KUNIMORI K, ICHIKUNI N, OKABE K, KUSAMA H, SAYAMA K, ARAKAWA H. CO2 hydrogenation activity and surface structure of zeolite-supported Rh catalysts[J]. Appl Catal A: Gen,1998,173(1):47−60.
JOO O-S, JUNG K-D, MOON I, ROZOVSKII A, LIN G, HAN S, UHM S. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the CAMERE Process) [J]. Ind Eng Chem Res, 1999, 38: 1808–1812.
张鲁湘, 张永春, 陈绍云, 助剂TiO2对CO2催化加氢制甲醇催化剂CuO-ZnO-Al2O3性能的影响 [J]. 燃料化学学报, 2011, 39(12): 912–917.ZHANG Lu-xiang, ZHANG Yong-chun, CHEN Shao-yun. Effect of promoter TiO2 on the performance of CuO-ZnO-Al2O3 catalyst for CO2 catalytic hydrogenation to methanol [J]. J Fuel Chem Technol, 2011, 39(12): 912–917.
ZHANG L, ZHANG Y, CHEN S. Effect of promoter SiO2, TiO2 or SiO2-TiO2 on the performance of CuO-ZnO-Al2O3 catalyst for methanol synthesis from CO2 hydrogenation [J]. Appl Catal A: Gen, 2012, 415–416: 118–123.
KOIZUMI N, JIANG X, KUGAI J, SONG C. Effects of mesoporous silica supports and alkaline promoters on activity of Pd catalysts in CO2 hydrogenation for methanol synthesis [J]. Catal Today, 2012, 194: 16–24.
STUDT F, SHARAFUTDINOV I, ABILD-PEDERSEN F, ELKJÆR C, HUMMELSHØJ J, DAHL S, CHORKENDORFF I, NØRSKOV J. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol [J]. Nat Chem, 2014, 6: 320–324.
XIAO J, MAO D, GUO X, YU J. Effect of TiO2, ZrO2, and TiO2-ZrO2 on the performance of CuO-ZnO catalyst for CO2 hydrogenation to methanol [J]. Appl Sur Sci, 2015, 338: 146–153.
CAI W, DE LA PISCINA P, TOYIR J, HOMS N. CO2 hydrogenation to methanol over CuZnGa catalysts prepared using microwave-assisted methods [J]. Catal Today, 2015, 242: 193–199.
KUSAMA H, OKABE K, SAYAMA K, ARAKAWA H. Ethanol synthesis by catalytic hydrogenation of CO2 over Rh-Fe/SiO2 catalysts [J]. Energy, 1997, 22(2/3): 343–348.
KUSAMA H, OKABE K, SAYAMA K, ARAKAWA H. CO2 hydrogenation to ethanol over promoted Rh/SiO2 catalysts [J]. Catal Today, 1996, 28: 261–266.
INUI T, YAMAMOTO T. Effective synthesis of ethanol from CO2 on polyfunctional composite catalysts [J]. Catal Today, 1998, 45: 209–214.
NAIK S, RYU T, BUI V, MILLER J, DRINNAN N, ZMIERCZAK W, Synthesis of DME from CO2/H2 gas mixture [J]. Chem Eng J, 2011, 167: 362–368.
BONURA G, CORDARO M, SPADARO L, CANNILLA C, ARENA F, FRUSTERI F. Hybrid Cu-ZnO-ZrO2/H-ZSM5 system for the direct synthesis of DME by CO2 hydrogenation[J]. Appl Catal B: Environ,2013,140–141:16−24.
WANG Y, WANG W, CHEN Y, MA J, LI R. Synthesis of dimethyl ether from syngas over core-shell structure catalyst CuO-ZnO-Al2O3@SiO2-Al2O3[J]. Chem Eng J,2014,250:248−256.
FRUSTERI F, CORDARO M, CANNILLA C, BONURA G. Multifunctionality of Cu-ZnO-ZrO2/H-ZSM5 catalysts for the one-step CO2-to-DME hydrogenation reaction[J]. Appl Catal B: Environ,2015,162:57−65.
SUO Z, KOU Y, NIU J, ZHANG W, WANG H. Characterization of TiO2-, ZrO2- and Al2O3-supported iron catalysts as used for CO2 hydrogenation[J]. Appl Catal A: Gen,1997,148:301−313.
LI C, YUAN X, FUJIMOTO K. Direct synthesis of LPG from carbon dioxide over hybrid catalysts comprising modified methanol synthesis catalyst and β-type zeolite[J]. Appl Catal A: Gen,2014,475:155−160.
FUJIWARA M, SOUMA Y. Hydrocarbon synthesis from carbon dioxide and hydrogen over Cu-Zn-Cr oxide/zeolite hybrid catalysts[J]. J Chem Soc, Chem Commun,1992,23(42):767−768.
LI C, FUJIMOTO K. Efficient conversion of carbon dioxide to non-methane light hydrocarbons-two stage process with intercooler[J]. Fuel Process Technol,2015,136:50−55.
AL-DOSSARY M, ISMAIL A, FIERRO J, BOUZID H, AL-SAYARI S. Effect of Mn loading onto MnFeO nanocomposites for the CO2 hydrogenation reaction[J]. Appl Catal B: Environ,2015,165:651−660.
FUJIWARA M, SAKURAI H, SHIOKAWA K, LIZUKA Y. Synthesis of C2+ hydrocarbons by CO2 hydrogenation over the composite catalyst of Cu-Zn-Al oxide and Hβ zeolite using two-stage reactor system under low pressure[J]. Catal Today,2015,242:255−260.
GAO P, DANG S, LI S, BU X, LIU Z, QIU M, YANG C, WANG H, ZHONG L, HAN Y, LIU Q, WEI W, SUN Y. Direct production of lower olefins from CO2 conversion via bifunctional catalysis[J]. ACS Catal,2018,8:571−578. doi: 10.1021/acscatal.7b02649
WANG S, WANG P, QIN Z, YAN W, DONG M, LI J, WANG J, FAN W. Enhancement of light olefin production in CO2 hydrogenation over In2O3-based oxide and SAPO-34 composite[J]. J Catal,2020,391:459−470. doi: 10.1016/j.jcat.2020.09.010
WANG X, ZENG C, GONG N, ZHANG T, WU Y, ZHANG J, SONG F, YANG G, TAN Y. Effective suppression of CO selectivity for CO2 hydrogenation to high-quality gasoline[J]. ACS Catal,2021,11:1528−1547. doi: 10.1021/acscatal.0c04155
WANG Y, TAN L, TAN M, ZHANG P, FANG Y, YONEYAMA Y, YANG G, TSUBAKI N. Rationally designing bifunctional catalysts as an efficient strategy to boost CO2 hydrogenation producing value-added aromatics[J]. ACS Catal,2019,9:895−901. doi: 10.1021/acscatal.8b01344
ZHANG J, LU S, SU X, FAN S, MA Q, ZHAO T. Selective formation of light olefins from CO2 hydrogenation over Fe-Zn-K catalysts[J]. J CO2 Util,2015,12:95−100. doi: 10.1016/j.jcou.2015.05.004
BURK M, LEE J, MARTINEZ J. A versatile tandem catalysis procedure for the preparation of novel amino acids and peptides[J]. J Am Chem Soc,1994,116:10847−10848.
BALEMA V, HEY-HAWKINS E. The CuCl-catalyzed reaction of trimethylsilyl(t-butyl) chlorophosphane with dimethylzirco-nocene: An example for tandem catalysis[J]. Z Anorg Allg Chem,1996,622:2053−2056. doi: 10.1002/zaac.19966221209
WILSON E. Heterogeneous tandem catalysis[J]. Chem Eng News,2011,89(16):9.
LOHR T, MARKS T. Orthogonal tandem catalysis[J]. Nat Chem,2015,7:477−482. doi: 10.1038/nchem.2262
CHENG K, GU B, LIU X, KANG J, ZHANG Q, WANG Y. Direct and highly selective conversion of synthesis gas to lower olefins: Design of a bifunctional catalyst combining methanol synthesis and carbon-carbon coupling[J]. Angew Chem Int Ed,2016,55:4725−4728. doi: 10.1002/anie.201601208
LIU X, ZHOU W, YANG Y, CHENG K, KANG J, ZHANG L, ZHANG G, MIN X, ZHANG Q, WANG Y. Design of efficient bifunctional catalysts for direct conversion of syngas into lower olefins via methanol/dimethyl ether intermediates[J]. Chem Sci,2018,9:4708−4718. doi: 10.1039/C8SC01597J
LI N, JIAO F, PAN X, CHEN Y, FENG J, LI G, BAO X. High-quality gasoline directly from syngas by dual metal oxide-zeolite (OX-ZEO) catalysis[J]. Angew Chem Int Ed,2019,58:1−6. doi: 10.1002/anie.201813481
CHENG K, ZHOU W, KANG J, HE S, SHI S, ZHANG Q, PAN Y, WEN W, WANG Y. Bifunctional catalysts for one-step conversion of syngas into aromatics with excellent selectivity and stability[J]. Chem,2017,3:334−347. doi: 10.1016/j.chempr.2017.05.007
ZHANG P, TAN L, YANG G, TSUBAKI N. One-pass selective conversion of syngas to para–xylene[J]. Chem Sci,2017,8:7941−7946. doi: 10.1039/C7SC03427J
XU Y, LIU J, WANG J, MA G, LIN J, YANG Y, LI Y, ZHANG C, DING M. Selective conversion of syngas to aromatics over Fe3O4@MnO2 and hollow HZSM-5 bifunctional catalysts[J]. ACS Catal,2019,9:5147−5156. doi: 10.1021/acscatal.9b01045
LI Z, WANG J, QU Y, LIU H, TANG C, MIAO S, FENG Z, AN H, LI C. Highly selective conversion of carbon dioxide to lower olefins[J]. ACS Catal,2017,7:8544−8548. doi: 10.1021/acscatal.7b03251
LI Z, QU Y, WANG J, LIU H, LI M, MIAO S, LI C. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts[J]. Joule,2019,3:570−583. doi: 10.1016/j.joule.2018.10.027
DANG S, LI S, YANG C, CHEN X, LI X, ZHONG L, GAO P, SUN Y. Selective transformation of CO2 and H2 into lower olefins over In2O3-ZnZrOx/SAPO-34 bifunctional catalysts[J]. ChemSusChem,2019,12:3582−3591. doi: 10.1002/cssc.201900958
GAO J, JIA C, LIU B. Direct and selective hydrogenation of CO2 to ethylene and propene by bifunctional catalysts[J]. Catal Sci Technol,2017,7:5602−5607. doi: 10.1039/C7CY01549F
LIU X, WANG M, ZHOU C, ZHOU W, CHENG K, KANG J, ZHANG Q, DENG W, WANG Y. Selective transformation of carbon dioxide into lower olefins with a bifunctional catalyst composed of ZnGa2O4 and SAPO-34[J]. Chem Commun,2018,54:140−143. doi: 10.1039/C7CC08642C
杨浪浪, 孟凡会, 张 鹏, 梁晓彤, 李 忠. ZrCdOx/SAPO-18双功能催化剂催化CO2加氢合成低碳烯烃性能 [J]. 无机化学学报, 2021, 37(3): 448–456.YANG Lang-lang, MENG Fan-hui, ZHANG Peng, LIANG Xiao-tong, LI Zhong, Catalytic performance for CO2 hydrogenation to light olefins Over ZrCdOx/SAPO-18 bifunctional catalyst [J]. Chin J Inorg Chem, 2021, 37(3): 448–456.
CHEN J, WANG X, WU D, ZHANG J, MA Q, GAO X, LAI X, XIA H, FAN S, ZHAO T-S. Hydrogenation of CO2 to light olefins on CuZnZr@(Zn-)SAPO-34 catalysts: Strategy for product distribution[J]. Fuel,2019,239:44−52. doi: 10.1016/j.fuel.2018.10.148
WANG X, YANG G, ZHANG J, CHEN S, WU Y, ZHANG Q, WANG J, HAN Y, TAN Y. Synthesis of isoalkanes over a core (Fe-Zn-Zr)-shell (zeolite) catalyst by CO2 hydrogenation[J]. Chem Commun,2016,52:7352−7355. doi: 10.1039/C6CC01965J
WANG X, YANG G, ZHANG J, SONG F, WU Y, ZHANG T, ZHANG Q, TSUBAKI N, TAN Y. Macroscopic assembly style of catalysts significantly determining their efficiency for converting CO2 to gasoline[J]. Catal Sci Technol,2019,9:5401−5412. doi: 10.1039/C9CY01470E
WEI J, SUN J, WEN Z, FANG C, GE Q, XU H. New insights into the effect of sodium on Fe3O4-based nanocatalysts for CO2 hydrogenation to light olefins[J]. Catal Sci Technol,2016,6:4786−4793. doi: 10.1039/C6CY00160B
WEI C, TU W, JIA L, LIU Y, LIAN H, WANG P, ZHANG Z. The evolutions of carbon and iron species modified by Na and their tuning effect on the hydrogenation of CO2 to olefins[J]. Appl Surf Sci,2020,525:146622.
ZHANG Z, WEI C, JIA L, LIU Y, SUN C, WANG P, TU W. Insights into the regulation of FeNa catalysts modified by Mn promoter and their tuning effect on the hydrogenation of CO2 to light olefins[J]. J Catal,2020,390:12−22.
ZHANG Y, CAO C, ZHANG C, ZHANG Z, LIU X, YANG Z, ZHU M, MENG B, XU J, HAN Y–F. The study of structure–performance relationship of iron catalyst during a full life cycle for CO2 hydrogenation[J]. J Catal,2019,378:51−62.
ZHANG Y, FU D, LIU X, ZHANG Z, ZHANG C, SHI B, XU J, HAN Y–F. Operando spectroscopic study of dynamic structure of iron oxide catalysts during CO2 hydrogenation[J]. ChemCatChem,2018,10:1272−1276.
HUANG J, JIANG S, WANG M, WANG X, GAO J, SONG C. Dynamic evolution of Fe and carbon species over different ZrO2 supports during CO prereduction and their effects on CO2 hydrogenation to light olefins[J]. ACS Sustainable Chem Eng,2021,9:7891−7903.
YUAN F, ZHANG G, ZHU J, DING F, ZHANG A, SONG C, GUO X. Boosting light olefin selectivity in CO2 hydrogenation by adding Co to Fe catalysts within close proximity[J]. Catal Today,2021,371:142−149.
LIU J, ZHANG G, JIANG X, WANG J, SONG C, GUO X. Insight into the role of Fe5C2 in CO2 catalytic hydrogenation to hydrocarbons[J]. Catal Today,2021,371:162−170.
LIU J, LI K, SONG Y, SONG C, GUO X. Selective hydrogenation of CO2 to hydrocarbons: effects of Fe3O4 particle size on reduction, carburization, and catalytic performance[J]. Energy Fuels,2021,35:10703−10709.
PHONGAMWONG T, CHANTAPRASERTPORN U, WITOON T, NUMPILAI T, POO-ARPORN Y, LIMPHIRAT W, DONPHAI W, DITTANET P, CHAREONPANICH M, LIMTRAKUL J. CO2 hydrogenation to methanol over CuO-ZnO-ZrO2-SiO2 catalysts: Effects of SiO2 contents[J]. Chem Eng J,2017,316:692−703. doi: 10.1016/j.cej.2017.02.010
MOU J, FAN X, LIU F, WANG X, ZHAO T, CHEN P, LI Z, YANG C, CAO J. CO2 hydrogenation to lower olefins over Mn2O3-ZnO/SAPO-34 tandem catalysts[J]. Chem Eng J,2021,421:129978.
YE J, LIU C, MEI D, GE Q. Active oxygen vacancy site for methanol synthesis from CO2 hydrogenation on In2O3(110): A DFT study[J]. ACS Catal,2013,3:1296−1306.
SUN K, FAN Z, YE J, YAN J, GE Q, LI Y, HE W, YANG W, LIU C. Hydrogenation of CO2 to methanol over In2O3 catalyst[J]. J CO2 Utili,2015,12:1−6.
WANG J, SUN K, JIA X, LIU C. CO2 hydrogenation to methanol over Rh/In2O3 catalyst[J]. Catal Today,2021,365:341−347.
WANG W, CHEN Y, ZHANG M. Facet effect of In2O3 for methanol synthesis by CO2 hydrogenation: A mechanistic and kinetic study[J]. Surf Interfaces,2021,25:101244.
DONG X, LI F, ZHAO N, XIAO F, WANG J, TAN Y. CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared by precipitation-reduction method[J]. Appl Catal B: Environ,2016,191:8−17. doi: 10.1016/j.apcatb.2016.03.014
CHEN S, ZHANG J, SONG F, ZHANG Q, YANG G, ZHANG M, WANG X, XIE H, TAN Y. Induced high selectivity methanol formation during CO2 hydrogenation over a CuBr2-modified CuZnZr catalyst[J]. J Catal,2020,389:47−59. doi: 10.1016/j.jcat.2020.05.023
FUJIWARA M, SOUMA Y. Hydrocarbon synthesis from carbon dioxide and hydrogen over Cu-Zn-Cr oxide/zeolite hybrid catalysts[J]. J Chem Soc, Chem Commun,1992,10:767−768.
PARK YK, PARK KC, IHM SK. Hydrocarbon synthesis through CO2 hydrogenation over CuZnOZrO2/zeolite hybrid catalysts[J]. Catal Today,1998,44:165−73. doi: 10.1016/S0920-5861(98)00187-4
WANG S, ZHANG L, ZHANG W, WANG P, QIN Z, YAN W, DONG M, LI J, WANG J, HE L, OLSBYE U, FAN W. Selective conversion of CO2 into propene and butene[J]. Chem,2020,6:3344−3363. doi: 10.1016/j.chempr.2020.09.025
TAN L, ZHANG P, CUI Y, SUZUKI Y, LI H, GUO L, YANG G, TSUBAKI N. Direct CO2 hydrogenation to light olefins by suppressing CO by-product formation[J]. Fuel Process Technol,2019,196:106174. doi: 10.1016/j.fuproc.2019.106174
MARTIN O, MARTIN A J, MONDELLI C, MITCHELL S, SEGAWA T F, HAUERT R, DROUILLY C, FERRE D C, RAMÍREZ J P. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation[J]. Angew Chem Int Ed,2016,55:6261−6265. doi: 10.1002/anie.201600943
XU Q, HE D, FUJIWARA M, SOUMA Y. Improved activity of Fe-Cu catalysts by physical mixing with zeolites for the hydrogenation of carbon dioxide[J]. J Mol Catal A: Chem,1997,120:L23−L26. doi: 10.1016/S1381-1169(97)00014-9
Xu Q, HE D, FUJIWARA M, TANAKA M, SOUMA Y, YAMANAKA H. Hydrogenation of carbon dioxide over Fe-Cu-Na/zeolite composite catalysts: Na migration via solid–solid reaction and its effects on the catalytic activity[J]. J Mol Catal A: Chem,1998,136:161−168. doi: 10.1016/S1381-1169(98)00047-8
WEI J, YAO R, GE Q, WEN Z, JI X, FANG C, ZHANG J, XU H, SUN J. Catalytic hydrogenation of CO2 to isoparaffins over Fe-based multifunctional catalysts[J]. ACS Catal,2018,8:9958−9967. doi: 10.1021/acscatal.8b02267
GENG S, JIANG F, XU Y, LIU X. Iron-based Fischer-Tropsch synthesis for the efficient conversion of carbon dioxide into isoparaffins[J]. ChemCatChem,2016,8:1303−1307. doi: 10.1002/cctc.201600058
NOREEN A, LI M, FU Y, AMOO C, WANG J, MATURURA E, DU C, YANG R, XING C, SUN J. One-pass hydrogenation of CO2 to multibranched isoparaffins over bifunctional zeolite-based catalysts[J]. ACS Catal,2020,10:14186−14194. doi: 10.1021/acscatal.0c03292
TAN Y, FUJIWARA M, ANDO H, XU Q, SOUMA Y. Syntheses of isobutane and branched higher hydrocarbons from carbon dioxide and hydrogen over composite catalysts[J]. Ind Eng Chem Res,1999,38:3225−3229. doi: 10.1021/ie980672m
BAI R, TAN Y, HAN Y. Study on the carbon dioxide hydrogenation to iso−alkanes over Fe-Zn-M/zeolite composite catalysts[J]. Fuel Process Technol,2004,86:293−301. doi: 10.1016/j.fuproc.2004.05.001
白荣献, 谭猗生, 韩怡卓. Fe-Zn-Zr/分子筛复合催化剂上CO2加氢合成异构烷烃Ⅰ不同分子筛对催化剂性能的影响[J]. 催化学报,2004,25(3):223−226. doi: 10.3321/j.issn:0253-9837.2004.03.013BAI Rong-xian, TAN Yi-sheng, HAN Yi-zhuo. Hydrogenation of carbon dioxide to isoalkanes over Fe-Zn-Zr/zeolite composite catalysts Ⅰ Effects of zeolites on catalytic performance of the catalysts[J]. Chin J Catal,2004,25(3):223−226. doi: 10.3321/j.issn:0253-9837.2004.03.013
NI X, TAN Y, HAN Y, TSUBAKI N. Synthesis of isoalkanes over Fe-Zn-Zr/HY composite catalyst through carbon dioxide hydrogenation[J]. Catal Commun,2007,8:1711−1714. doi: 10.1016/j.catcom.2007.01.023
WEI J, GE Q, YAO R, WEN Z, FANG C, GUO L, XU H, SUN J. Directly converting CO2 into a gasoline fuel[J]. Nat Commun,2017,8:15174−15181. doi: 10.1038/ncomms15174
GAO P, LI S, BU X, DANG S, LIU Z, WANG H, ZHONG L, QIU M, YANG C, CAI J, WEI W, SUN Y. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst[J]. Nat Chem,2017,9:1019−1024. doi: 10.1038/nchem.2794
CUI X, GAO P, LI S, YANG C, LIU Z, WANG H, ZHONG L, SUN Y. Selective production of aromatics directly from carbon dioxide hydrogenation[J]. ACS Catal,2019,9:3866−3876. doi: 10.1021/acscatal.9b00640
XU Y, SHI C, LIU B, WANG T, ZHENG J, LI W, LIU D, LIU X. Selective production of aromatics from CO2[J]. Catal Sci Technol,2019,9:593−610. doi: 10.1039/C8CY02024H
WANG S, WU T, LIN J, TIAN J, JIY, PEI Y, YAN S, QIAO M, XU H, ZONG B. FeK on 3D graphene-zeolite tandem catalyst with high efficiency and versatility in direct CO2 conversion to aromatics[J]. ACS Sustainable Chem Eng,2019,7:17825−17833. doi: 10.1021/acssuschemeng.9b04328
SONG G, LI M, YAN P, ASIF NAWAZ M, LIU D. High conversion to aromatics via CO2-FT over a CO-reduced Cu-Fe2O3 catalyst integrated with HZSM-5[J]. ACS Catal,2020,10:11268−11279. doi: 10.1021/acscatal.0c02722
ZHOU C, SHI J, ZHOU W, CHENG K, ZHANG Q, KANG J, WANG Y. Highly active ZnO-ZrO2 aerogels integrated with H-ZSM-5 for aromatics synthesis from carbon dioxide[J]. ACS Catal,2020,10:302−310. doi: 10.1021/acscatal.9b04309
ZHANG X, ZHANG A, JIANG X, ZHU J, LIU J, LI J, ZHANG G, SONG C, GUO X. Utilization of CO2 for aromatics production over ZnO/ZrO2-ZSM-5 tandem catalyst[J]. J CO2 Util,2019,29:140−145. doi: 10.1016/j.jcou.2018.12.002
NI Y, CHEN Z, FU Y, LIU Y, ZHU W, LIU Z. Selective conversion of CO2 and H2 into aromatics[J]. Nat Commun,2018,9:3457−3463. doi: 10.1038/s41467-018-05880-4
ZHANG J, ZHANG M, CHEN S, WANG X, ZHOU Z, WU Y, ZHANG T, YANG G, HAN Y, TAN Y. Hydrogenation of CO2 into aromatics over a ZnCrOx-zeolite composite catalyst[J]. Chem Commun,2019,55:973−976. doi: 10.1039/C8CC09019J
WANG Y, GAO W, KAZUMI S, LI H, YANG G, TSUBAKI N. Direct and oriented conversion of CO2 into value-added aromatics[J]. Chem Eur J,2019,25:5149−5153. doi: 10.1002/chem.201806165

图 1 CO2加氢制备C2+烃的串联催化过程
Figure 1 Tandem catalysis of CO2 hydrogenation into C2+ hydrocarbons

图 2 不同串联催化剂上CO2加氢制低碳烯烃的可能反应机制
Figure 2 Possible reaction mechanisms for CO2 hydrogenation to lower olefins on different tandem catalysts
(a): Zn-Ga-O/SAPO-34[43]; (b): ZnZrO/SAPO[39] reproduced with permission from ref.[43, 39], Copyright (2018) The Royal Society of Chemistry, Copyright (2017) American Chemical Society

图 3 (a) In-Zr及SAPO-34的组装方式对串联催化剂上CO2加氢制低碳烯烃性能的影响[24];(b) ZnZrO及SAPO的组装方式对串联催化剂上CO2加氢制低碳烯烃性能的影响[39];(c) CuZnZr及SAPO-34两组分间的界面示意图[45]
Figure 3 (a) Effect of the assembly style between In-Zr and SAPO-34 on CO2 hydrogenation to lower olefins[24]; (b) Effect of the assembly style between ZnZrO and SAPO on CO2 hydrogenation to lower olefins[39]; (c) Schematic diagram of the interface between CuZnZr and SAPO-34[45] reproduced with permission from ref.[24, 39, 45], Copyright (2018 & 2017) American Chemical Society, Copyright (2019) Elsevier


图 5 (a) In-Zr/SAPO-34的催化性能随原料气中CO浓度的变化趋势[24];(b)不同原料气氛下In2O3/ZrO2&SAPO催化剂上CO2转化制备低碳烯烃[68]
Figure 5 (a) Catalytic performance over In-Zr/SAPO-34 as a function of CO concentration[24]; (b) CO2 conversion to lower olefins from different feed gas over In2O3/ZrO2&SAPO catalyst[68] reproduced with permission from ref. [24, 68], Copyright (2018) American Chemical Society, Copyright (2019) Elsevier

图 6 不同串联催化剂上CO2加氢制低碳烯烃的(a)、(c)、(d)稳定性及(b)抗硫性能测试
Figure 6 (a), (c), (d) Stability and (b) sulfur tolerance test of different tandem catalysts for CO2 hydrogenation to lower olefins
(a), (b): 20%Mn2O3-ZnO/SAPO-34[58]; (c): In2O3-ZnZrOx/SAPO-34-C[41]; (d): In2O3-ZnZrOx/SAPO-34-H-a[41] reproduced with permission from ref. [58, 41], Copyright (2021) Elsevier, Copyright (2019) John Wiley and Sons

图 7 (a) Na-Fe3O4/HMCM-22串联催化剂上CO2加氢制异构烷烃及积炭形成的反应机制[72];(b) Fe-Zn-Zr/HY串联催化剂上CO2加氢制异构烷烃的反应路径[78];(c) 物理黏接法制备Fe-Zn-Zr@zeolite核壳催化剂的示意图[46]
Figure 7 (a) Reaction scheme of isoalkanes synthesis and coke formation during CO2 hydrogenation over Na-Fe3O4/HMCM-22 catalyst[72]; (b) Proposed reaction path of isoalkanes formation from CO2 hydrogenation over Fe-Zn-Zr/HY composite catalyst[78]; (c) Illustration for the Fe-Zn-Zr@zeolite core-shell catalyst preparation by a cladding method[46] reproduced with permission from ref. [72, 78, 46], Copyright (2018) American Chemical Society, Copyright (2007) Elsevier, Copyright (2016) The Royal Society of Chemistry

图 8 两类活性组分间的组装方式对不同串联催化剂上CO2加氢制汽油性能的影响
Figure 8 Effect of the assembly style between the two active sites on CO2 hydrogenation to gasoline over the different tandem catalysts
(a): Na-Fe3O4/HZSM-5[79]; (b): In2O3/HZSM-5[80]; (c): Fe-Zn-Zr@HZSM-5[47] reproduced with permission from ref. [79, 80, 47], Copyright (2017) Springer Nature, Copyright (2019) The Royal Society of Chemistry

图 9 不同串联催化剂上CO2加氢制(a)、(b)异构烷烃及(c)、(d)汽油的稳定性测试
Figure 9 Stability tests of the different tandem catalysts for CO2 hydrogenation to (a), (b) isoalkanes and (c), (d) gasolines
(a): Fresh Na-Fe3O4/zeolite[72]; (b): Regenerated Na-Fe3O4/HMCM-22[72]; (c): Na-Fe3O4/HZSM-5[79]; (d): In2O3/HZSM-5[80] reproduced with permission from ref. [72, 79, 80], Copyright (2018) American Chemical Society, Copyright (2017) Springer Nature

图 10 不同串联催化剂上CO2加氢制芳烃的反应路径及机制
Figure 10 Reaction paths and schematics of CO2 hydrogenation to aromatics on different tandem catalysts
(a): ZnFeOx-4.25Na/S-HZSM-5[81]; (b): 6.25Cu-Fe2O3/HZSM-5[84]; (c); ZnZrO/HZSM-5[40] reproduced with permission from ref. [81, 84, 40], Copyright (2019 & 2020) American Chemical Society, Copyright (2019) Elsevier

图 11 (a)、(b)、(c) 组装方式对不同串联催化剂上CO2加氢制芳烃性能的影响;(d) FeK1.5/HSG|HZSM-5(50)串联催化剂上CO2转化制芳烃的烯化-芳构化反应路径
Figure 11 (a), (b), (c) Effect of the assembly style between the two active sites on CO2 hydrogenation to aromatics over the different tandem catalysts; (d) Illustration of the olefination-aromatization reaction pathway for converting CO2 to aromatics over the FeK1.5/HSG|HZSM-5(50) tandem catalyst
(a): ZnFeOx-4.25Na/S-HZSM-5[81]; (b): 6.25Cu-Fe2O3/HZSM-5-c[84]; (c), (d): FeK1.5/HSG-HZSM-5(50)[83] reproduced with permission from ref. [81, 84, 83], Copyright (2019 & 2020 & 2019) American Chemical Society

图 12 两类活性组分间的组装方式对不同串联催化剂上CO2加氢制芳烃性能的影响
Figure 12 Effect of the assembly style between two active sites on CO2 hydrogenation to aromatics over different tandem catalysts
(a): ZnZrO/HZSM-5[40]; (b): Cr2O3/HZSM-5[27]; (c): ZnAlOx&HZSM-5[87] reproduced with permission from ref. [40, 27, 87], Copyright (2019) Elsevier, Copyright (2019) American Chemical Society

图 13 (a) 分子筛类型对FeK1.5/HSG|zeolite催化剂上CO2转化率及产物选择性的影响[83];(b) 串联催化剂上芳烃分布及 (c)Cr2O3/H-ZSM-5@S-1上高选择性生成BTX的机制[27]
Figure 13 (a) Effect of zeolite types on the CO2 conversion and product selectivity over the FeK1.5/HSG|zeolite catalysts[83]; (b) Fractions of products in aromatics over the tandem catalysts and (C) Scheme of highly selective production of BTX over Cr2O3/H-ZSM-5@S-1[27] reproduced with permission from ref. [83, 27], Copyright (2019) American Chemical Society

图 14 (a) N2及CO2气氛下HZSM-5及Cr2O3/H-ZSM-5催化剂上甲醇芳构化性能及(b)串联反应中合成芳烃的CO2-辅助作用示意图[89]
Figure 14 (a) Catalytic performances of H-ZSM-5 and Cr2O3/H-ZSM-5 for MTA under N2 and CO2 atmospheres and (b) Schematic representation of the CO2-assisted effect for the synthesis of aromatics in the tandem reaction[89] reproduced with permission from ref. [89], Copyright (2019) John Wiley and Sons

图 15 不同串联催化剂上CO2加氢制芳烃的稳定性测试
Figure 15 Stability tests of the different tandem catalysts for CO2 hydrogenation to aromatics
(a) ZnZrO/ZSM-5[40]; (b) ZnAlOx&H-ZSM-5[87]; (c) ZnO/ZrO2-ZSM5[86]; (d) ZnFeOx-4.25Na/S-HZSM-5[81] reproduced with permission from ref. [40, 87, 86, 81], Copyright (2019 & 2019) Elsevier, Copyright (2019) American Chemical Society

图 16 CO2加氢制备C2+烃的串联催化过程
Figure 16 Tandem catalysis process of CO2 hydrogenation to C2+ hydrocarbons
表 1 串联催化剂上CO2加氢制低碳烯烃的催化性能
Table 1. Catalytic performances of CO2 hydrogenation to lower olefins over the tandem catalysts
| Catalyst | t /℃ | p /MPa | | |
| In-Zr/SAPO-34 (Granule-mixing)[24] | 400 | 3 | 35.5 | 76.4 |
| In2O3-ZnZrOx/SAPO-34 (Granule-mixing)[41] | 380 | 3 | 17.0 | 85.0 |
| 1In2O3/ZrO2-1SAPO-34 (Powder-mixing)[42] | 400 | 1.5 | ~20.0 | 80.0–90.0 |
| InCrOx(0.13)/SAPO-34 (Powder-mixing)[25] | 350 | 1 | 17.2 | 89.1 |
| ZnGa2O4/SAPO-34 (Powder-mixing)[43] | 370 | 3 | 13.0 | 86.0 |
| ZnZrO/SAPO-34 (Powder-mixing)[39] | 380 | 2 | 12.6 | 80.0 |
| Zr8Cd1/SAPO-18 (Powder-mixing)[44] | 370 | 2.5 | 17.8 | 85.6 |
| CuZnZr@Zn–SAPO-34 (Core-shell)[45] | 400 | 2 | 19.6 | 60.5 |
| Zn0.5Ce0.2Zr1.8O4/H-RUB-13 (Powder-mixing)[57] | 350 | 1 | 10.7 | 83.4 |
| Mn2O3-ZnO/SAPO-34 (Ball milling-mixing)[58] | 380 | 3 | 29.8 | 80.2 |
| *: | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 串联催化剂上CO2加氢制异构烷烃及汽油的催化性能
Table 2. Catalytic performances of CO2 hydrogenation to isoalkanes and gasoline over the tandem catalysts
| Catalyst | t /℃ | p /MPa | | sisoalkanes or gasoline /% |
| Fe-Cu-Na/US-Y(10.7) (Physical-mixing)[71] | 250 | 2 | 11.5 | 77.3a |
| Na-Fe3O4/HMCM-22(Dual-bed)[72] | 320 | 3 | 26.0 | 74.0b |
| 92.6Fe7.4K+HZSM-5 (Granule-mixing)[73] | 300 | 2.5 | 43.9 | 69.7a |
| NaFe+SAPO-11+HZSM-5(Triple-bed)[74] | 320 | 3 | 31.2 | 38.2c |
| Fe-Zn-Zr/HY (Granule-mixing)[76] | 340 | 5 | 22.4 | 55.3a |
| Fe-Zn-Zr@HZSM-5-Hbeta (Core-shell)[46] | 340 | 5 | 14.9 | 81.3d |
| Fe-Zn-Zr@HZSM-5 (Core-shell)[47] | 340 | 5 | 21.5 | 91.9e |
| Na-Fe3O4/HZSM-5 (Granule-mixing)[79] | 320 | 3 | 22.0 | 78.0f |
| In2O3/HZSM-5 (Granule-mixing)[80] | 340 | 3 | 13.1 | 78.6f |
| a: C4–C6 isoalkanes selectivity in C4–C6 hydrocarbons;b: C4+ isoalkanes selectivity in C4+ hydrocarbons;c: C5+ isoalkanes selectivity in all hydrocarbons;d: C4+ isoalkanes selectivity in all hydrocarbons;e: C5+ isoalkanes selectivity in C5+ hydrocarbons;f: C5–C11 or C5+ hydrocarbons in all hydrocarbons | ||||
下载: 导出CSV
表 3 串联催化剂上CO2加氢制芳烃的催化性能
Table 3. Catalytic performances of CO2 hydrogenation to aromatics over the tandem catalysts
| Catalyst | t /℃ | p /MPa | | saromatics /% |
| ZnFeOx-4.25Na/S-HZSM-5 (Granule-mixing)[81] | 320 | 3 | 41.2 | 75.6 |
| Na/Fe-HZSM-5 (Granule-mixing)[82] | 320 | 1 | 29.4 | 54.3 |
| FeK1.5/HSG-HZSM-5 (Dual-bed)[83] | 340 | 2 | 35.0 | 68.0 |
| 6.25Cu-Fe2O3/HZSM-5 (Granule-mixing)[84] | 320 | 3 | 57.3 | 56.6 |
| ZnZrO/ZSM-5 (Powder-mixing)[40] | 320 | 4 | 14.0 | 73.0 |
| ae-ZnO-ZrO2/Z5 (Powder-mixing)[85] | 340 | 4 | 16.0 | 76.0 |
| ZnO/ZrO2-ZSM-5 (Powder-mixing)[86] | 340 | 3 | 9.0 | 70.0 |
| ZnAlOx & HZSM-5 (Powder-mixing)[87] | 320 | 3 | 9.1 | 73.9 |
| Cr2O3/HZSM-5 (Powder-mixing)[27] | 350 | 3 | 34.5 | 76.0 |
| ZnCrOx-ZnZSM-5 (Powder-mixing)[88] | 320 | 5 | 19.9 | 81.1* |
| * Aromatics selectivity in C5+ hydrocarbons, the others are the aromatics selectivity in all hydrocarbons | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
