图 1.
Ru-Co3O4的SEM((a)、(b))和TEM照片(c)
Figure 1.
SEM images ((a), (b)) and TEM images (c) of Ru-Co3O4

含碳资源的利用会产生大量二氧化碳(CO2)。这些CO2排放到大气中,造成了一系列环境问题,如全球气温上升、海洋酸化等[1]。将CO2通过加氢的方式绿色高效地催化转化为有价值的化学品,不仅具有重要的价值,而且对于能源储存、CO2减排和可持续社会模式的建立也具有重要的意义[2]。甲烷(CH4)分子作为载能分子,可以高效地储存氢能,而且可以利用现有天然气基础设施进行安全的运输和使用,因此,将CO2催化加氢制CH4是CO2转化利用的一条路线,受到了研究者的广泛关注[3]。
目前,CO2催化加氢制CH4的反应过程分为气相和液相两种。其中,气相加氢反应方面的研究较多,也取得了重要进展[4]。在气相反应过程中,为了获得较高的CO2转化率(60%−100%),反应温度通常较高,常在250−450 °C;同时为了抑制逆水煤气反应(CO2+H2→CO+H2O)的活性和副产物CO的选择性,反应常在常压或接近常压的条件下进行,催化剂的甲烷时空收率往往不高[5-7]。相比之下,液相CO2加氢制CH4在较低反应温度下(160−200 °C)就可以进行。在液相反应中,研究者多采用有机配体修饰的贵金属均相体系为催化剂;尽管这些催化剂表现出较高的本征活性,但催化剂成本高且分离和回收利用困难等问题限制了其大规模的商业化应用[8-10]。开发负载型贵金属多相催化剂体系有利于解决上述问题。但目前关于贵金属多相催化剂的液相CO2加氢反应的研究报道较少,这主要是由于贵金属多相催化剂的转化率不高。如Dorner等[11]研究了Co-Pt/Al2O3催化剂的液相CO2加氢活性;他们发现在220 °C、1.9 MPa、H2/CO2 = 1∶1的条件下,甲烷的选择性可达93%,但CO2的转化率较低,为6.8%。因此,提高贵金属多相催化剂的转化率是目前液相CO2加氢制CH4的挑战之一。
在液相反应体系中,除了催化剂之外,溶剂也是重要的组成部分;溶剂对催化反应的影响规律和作用本质,即溶剂效应,是液相催化反应体系的基本科学问题之一,受到研究者的普遍关注[12]。在液相CO2加氢反应中,人们发现溶剂对反应活性影响很大。如He等[13]采用Pt/Co3O4催化剂,深入研究了水这一溶剂在液相CO2加氢生成C2+醇类的作用本质;他们通过D2O和13CH3OH同位素标记实验发现,溶剂水通过参与加氢反应,降低了反应所需的活化能,从而降低了反应所需的温度。Filonenko等[14]研究了负载Au催化剂催化的液相CO2加氢制甲酸反应过程中的溶剂效应,发现甲酸的产率随着溶剂极性增加而增加。对于液相CO2加氢制CH4反应过程,如果能深入理解溶剂效应本质,不仅有望提高反应活性,而且对设计高效的催化反应体系也具有指导意义。但目前关于液相CO2加氢制CH4反应过程中的溶剂效应,还极少见文献报道。
鉴于此,本研究以液相CO2加氢制CH4反应为反应体系,以多相Ru-Co3O4体系为催化剂,以不同的溶剂(水、正丁醇、丁内酯、DMF、正壬烷、异辛烷、环己烷、十氢萘等)为反应体系,结合同位素标记实验和原位红外光谱,深入探究溶剂的作用本质。研究发现,以异辛烷和十氢萘为溶剂时,Ru-Co3O4的CO2转化率活性最高,分别可达42%和45.6%,甲烷选择性分别可达98.2%和97%。氘代实验发现,这两种溶剂促进催化剂活性的本质是它们可作为活性氢的载体,起到供氢的作用,从而促进加氢反应;相比伯碳和仲碳,叔碳具有更高的供氢能力。
实验所用试剂如下:硝酸钴(Co(NO3)2·6H2O,国药集团化学试剂有限公司),碳酸钠(Na2CO3,科密欧化学试剂有限公司),氯化钌(RuCl3,阿拉丁试剂有限公司),白炭黑(SiO2,阿拉丁试剂有限公司),氧化锆(ZrO2,阿拉丁试剂有限公司),氧化铈(CeO2,阿拉丁试剂有限公司),P25(TiO2,ACROS ORCANICS公司),正丁醇(C4H10O,国药集团化学试剂有限公司),1,4-丁内酯(C4H6O2,阿拉丁试剂有限公司),正壬烷(C9H20,杭州炼油厂),十氢萘(C10H18,阿拉丁试剂有限公司),异辛烷(C8H18,天津市福晨化学试剂厂),N,N-二甲基甲酰胺(DMF,北京北化精细化学品有限责任公司),环己烷(C6H12,阿拉丁试剂有限公司),以上试剂均为分析纯。
Ru-Co3O4催化剂采用共沉淀法制备,Ru负载量为0.96%(质量分数),与理论负载量1%相似[13]。具体制备方法如下:将1.5 g的Co(NO3)2·6H2O和8.5 mg的RuCl3溶解在20 g的蒸馏水中,然后在30 °C下20 min内将溶液滴加到100 mL的0.5 mol/L Na2CO3溶液中,继续搅拌1 h;沉淀物通过离心分离得到,并用蒸馏水反复洗涤,直到硝酸银(0.1 mol/L)溶液检测不到白色沉淀。然后,将所得的沉淀在80 °C干燥过夜,再在400 °C焙烧6 h,得到Ru-Co3O4催化剂。
Ru/SiO2、Ru/ZrO2、Ru/TiO2和Ru/CeO2催化剂(Ru的负载量为1%(质量分数))采用湿浸渍法制备。将5 g载体加到10 mL 50 mmol/L 的RuCl3水溶液中,在室温下搅拌2 h;所得混合物在80 °C干燥过夜,再在300 °C焙烧2 h。
X射线粉末衍射(XRD)谱图在日本Rigaku公司的Mini Flex II X衍射仪上获得。仪器采用Cu Kα辐射(λ = 0.15406 nm),管电压30 kV,管电流15 mA,5°-80°扫描,扫描速率为8(°)/min。
扫描电子显微镜(SEM)测试在日本电子株式会社的JSM-6700F型场发射电子显微镜上进行。
透射电子显微镜(TEM)在日本电子株式会社JEM -2010高分辨透射电子显微镜上采集。
核磁共振氢谱(1H-NMR)是在布鲁克AV-Ⅲ400 MHz 液体核磁共振波谱仪上测得的。该液体核磁共振波谱仪配备一个5 mm BB/19F-1H/D 探头。1H 共振频率为400.13 MHz。
气相色谱-质谱分析(GCMS)是在Shimadzu GCMS-QP2010Ultra气相色谱-质谱联用仪(配有Rtx-5MS毛细管柱(60 m × 0.25 mm × 0.25 μm))上进行。
原位漫反射红外光谱图在 Nicolet iS10 FT-IR 光谱仪上采集,扫描的波长为 4000–650 cm−1,扫描64次,分辨率为4 cm−1。在十氢萘的吸附实验中,将一定量的催化剂粉末置于原位反应池中,以氩气将十氢萘鼓泡带入原位池中直至十氢萘吸附饱和,然后室温下采集光谱,随后再通入氩气在室温下对原位池进行吹扫,吹扫完毕后采集光谱;在CO2原位升温反应实验中,将一定量的催化剂粉末置于原位反应池中,在室温下通入氩气吹扫,并采集谱图,作为背景;然后将溶剂滴加在催化剂上,通入CO2,使用加热控温装置控制样品池的温度,采集不同温度下的红外光谱。
程序升温还原(H2-TPR)在Micromeritics公司的 AutoChemⅡ2920 型多用吸附仪上进行。以Ar为载气,H2+Ar(H2体积分数10%)的混合气作为还原气,气体进入吸附仪前经过脱水、脱有机杂质、脱氧等多级净化处理,热导检测器(TCD)监测耗氢量。样品装量 50 mg(粉末),首先在300 °C下用Ar(30 mL/min)吹扫30 min,随后Ar气氛中冷却至室温,切换为还原气,待TCD信号稳定后,以10 °C/min 的速率从室温升至600 °C,记录程序升温过程中的TCD信号。
CO脉冲吸附在Micromeritics公司的 AutoChemⅡ2920 型多用吸附仪上进行。以Ar为载气,H2+Ar(H2体积分数10%)的混合气作为还原气,气体进入仪器前经过脱水、脱有机杂质、脱氧等多级净化处理,热导检测器(TCD)检测气体消耗量。样品填装量100 mg(粉末)。首先在100 °C的还原气氛(30 mL/min)中还原1 h;然后经Ar吹扫1 h降温至30 °C,恒温条件下脉冲注入一氧化碳气体至吸附至饱和。
催化剂的性能评价是在30 mL的高压反应釜中进行(带有聚四氟乙烯内衬)。反应前,将0.1 g的催化剂粉末在300 °C、H2/Ar混合气(H2和Ar的体积比为1∶9,60 mL/min)中的管式炉中还原2 h;然后,在室温下加入到含有5 g溶剂的反应釜中,并将4 MPa的H2/CO2混合气(H2∶CO2 = 3∶1,物质的量比)充入反应釜中待温度升至200 °C时,反应开始计时。在所有反应中,反应时间均为1 h。
待反应结束后,将反应釜放置在冰水浴中,快速冷却至室温;然后用2 L的气袋将反应后的气体收集起来;尾气的气体组成采用Agilent 7890A气相色谱仪分析,该气相色谱仪配有一个热导检测器(TCD)和两个火焰离子化检测器(FID),其中,H2、CO2、CO和CH4由TCD检测器检测(5A分子筛柱,3 m × 1/16, 80/100 目),CH4和C2−6烃类由一个FID检测器检测(Al2O3毛细管柱,50 m × 0.530 mm × 15.0 μm),另外一个FID检测器配备GS-OxyPLOT毛细管柱(10 m × 0.530 mm × 10.0 μm),用来检测含氧类产物(如甲醇、乙醇和二甲醚等)和芳烃类产物(如苯、甲苯、二甲苯和三甲苯等)。在所有的反应中,气相产物中均未检测到含氧类产物和芳烃类产物。
溶解度实验同样是在30 mL的高压反应釜中进行。在25 °C下,将12.5 mL的溶剂加入反应釜中,然后将3 MPa的H2充入反应釜中,密封,确保不漏气;为了准确测得反应釜的压力变化,采用灵敏度为1 kPa的数字显示压力表来计量压力变化;反应釜压力不再发生变化时的值记为p1,25 °C下溶剂的饱和蒸气压记为p2,氢气溶解于溶剂中引起的压力变化 = 3 MPa − ( p1 − p2)。氢气在溶剂中的溶解度按标况下理想气体方程计算n = Δpv/(8.314 × 298.15)。
氘代标记实验同样是在30 mL的高压反应釜中进行。将0.1 g的Ru-Co3O4催化剂粉末在300 °C下及H2/Ar混合气气氛(H2和Ar的体积比为1∶9,60 mL/min)中的管式炉中还原2 h;随后于室温下加入到含有5 g溶剂(正壬烷、十氢萘或者异辛烷)的反应釜中,然后充入D2直到压力达到1 MPa,密封,确保不漏气;将反应釜温度从室温升至200 °C,并在200 °C下分别保持3 h(正壬烷、十氢萘)和12 h(异辛烷)。然后,将反应釜放置在冰水浴中,快速冷却至室温;取出溶剂进行GC-MS和核磁共振氢谱分析。
十氢萘和异辛烷的氘代实验中,将87.1 mg吡嗪溶解在1 mL氘代氯仿作为内标物,每次取200 μL内标和200 μL样品混合进行评价。通过内标法来计算不同位置氢的氘代率。
每次反应之前将反应釜浸没在水中保证不漏气的情况下再进行反应。
CO2转化率和各组分选择性的计算方法如下:
CO2转化率(%)= 生成物对应的C(mol)/[生成物对应的C(mol)+ 反应后剩余CO2(mol)] × 100% (1)
烃类选择性(s)= 产物中某组分所含碳摩尔数/总产物中碳摩尔数 (2)
图1为Ru-Co3O4催化剂的SEM和TEM照片,在新鲜Ru-Co3O4催化剂中,新鲜样品Co3O4的晶粒堆叠团聚,形成较大颗粒,没有观察到Ru晶粒。同时,一氧化碳脉冲化学吸附实验[15-17]测得Ru分散度为12.2%,说明Ru高分散在Co3O4中。XRD表征结果(图2)进一步证明了这一点,新鲜样品的XRD谱图与单纯Co3O4的一致,没有Ru的特征峰,31.5°、36.8°、44.8°、59.6°和65.3°处的衍射峰分别对应Co3O4的(220)、(311)、(400)、(511)和(440)晶面[18]。
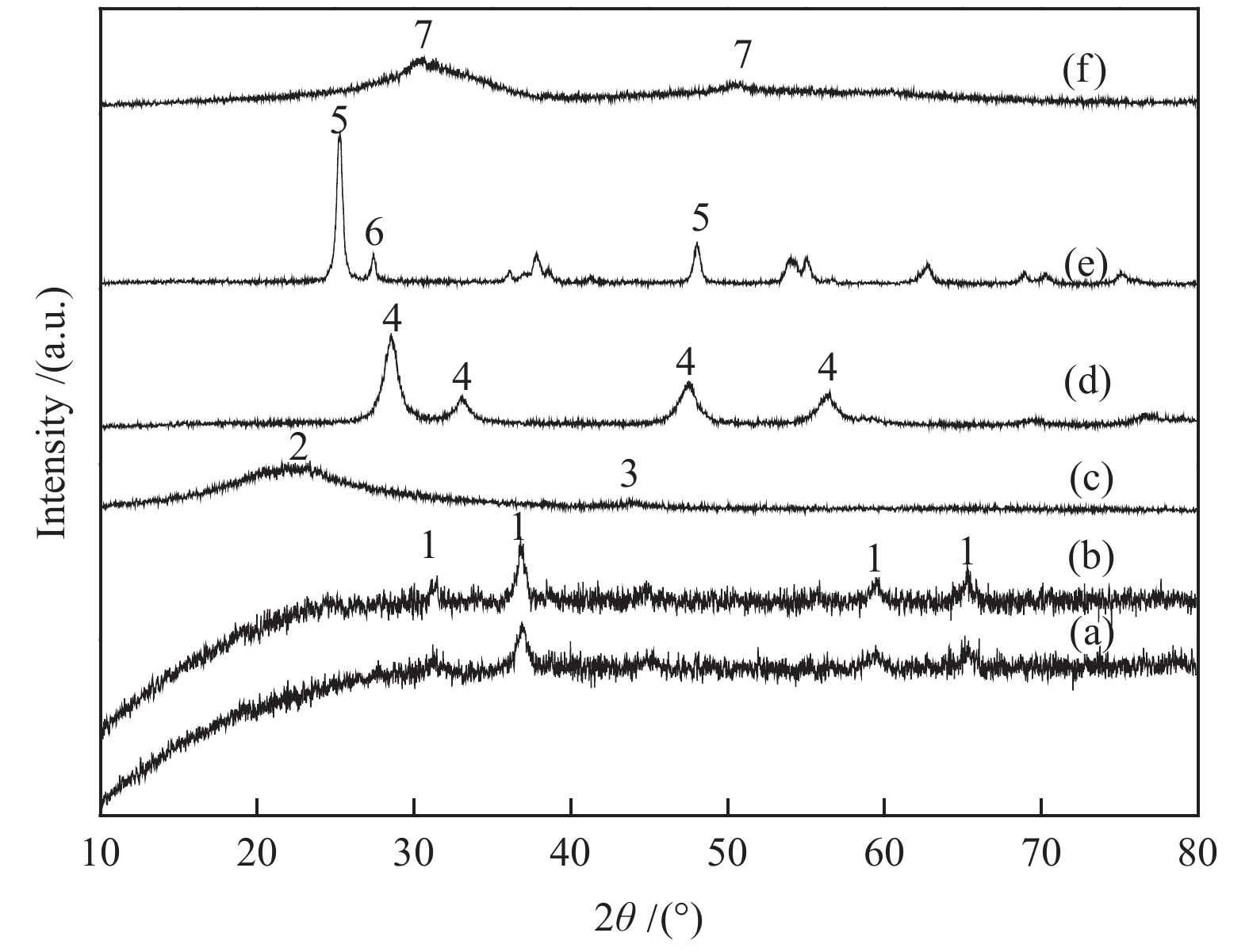
1: Co3O4; 2: SiO2; 3: Ru; 4: CeO2; 5, 6: the characteristic peaks of TiO2 with different crystal forms (5 is auatase and 6 is rutile); 7: ZrO2
在200 °C、4 MPa(H2/CO2 = 3∶1)的反应条件下,反应1 h后,Ru-Co3O4的CH4选择性为97%,CO2转化率为45.6%。相比较而言,Co-Pt/Al2O3,在220 °C、1.9 MPa、H2/CO2 = 1∶1的条件下,其CO2转化率不到10%[11]。由图2可知,Ru在SiO2、CeO2、ZrO2和TiO2载体上的分散度也很高,只有Ru/SiO2在44°处出现一个不明显的包峰[19]。由表1可知,载体对反应活性影响显著。Ru/SiO2催化剂的CO2加氢反应活性最低,CO2转化率仅为2.3%;Ru/CeO2催化剂的活性也较低,CO2转化率仅为5.4%;Ru/ZrO2和Ru/TiO2催化剂的活性达到11%−15%。载体种类对产物的选择性影响不大,不同催化剂的CH4选择性在97%−99%。除CH4外,还有少量C2−5的烃类生成。未检测到含氧化合物如甲醇、二甲醚、乙醇等产物。
 下载:
导出CSV
下载:
导出CSV
| Entry | Catalyst | CO2 conversion /% |
Selectivity of
hydrocarbons/mol% |
|||
| CH4 | C2−5 | CO | ||||
| 1 | Ru/SiO2 | 2.3 | 97.1 | 2.9 | 0 | |
| 2 | Ru/CeO2 | 5.4 | 98.8 | 1.2 | 0 | |
| 3 | Ru/ZrO2 | 11.3 | 97.9 | 2.1 | 0 | |
| 4 | Ru/TiO2 | 15.2 | 97.1 | 2.9 | 0 | |
| 5 | Ru-Co3O4 | 45.6 | 97.0 | 2.9 | 0.1 | |
| 6 | Co3O4 | 34.0 | 97.7 | 2.2 | 0.1 | |
| reaction conditions: 100 mg of catalyst, 5 g decalin, 200 °C, initial pressure: 4 MPa of H2/CO2 gas (v/v = 3∶1), 1 h | ||||||
鉴于Ru-Co3O4催化剂显示出优良的催化性能,下面以Ru-Co3O4催化剂来研究不同溶剂对反应性能的影响规律。表2为水、正丁醇、1,4-丁内酯、二甲基甲酰胺(DMF)、正壬烷、异辛烷和十氢萘溶剂中,Ru-Co3O4的催化反应结果。不同溶剂中Ru-Co3O4催化剂上的主要加氢产物均为CH4,其选择性在87%−99%;除CH4外,还有少部分C2−5烃类产物的产生。
下载:
导出CSV
| Solvent | CO2 conversion/% | Product selectivity/% | Solubility b/(mmol·L−1) | |
| CH4 | C2−5 | |||
| Water | 8.3 | 88.7 | 11.0 | 28.9 |
| Butyl alcohol | 21.4 | 88.7 | 11.1 | 87.2 |
| 1,4- butyrolactone | 26.2 | 95.7 | 4.2 | 42.6 |
| DMF | 31.0 | 89.4 | 10.4 | 43.2 |
| n-Nonane | 31.9 | 98.5 | 1.5 | 93.5 |
| Decalin | 45.6 | 97.0 | 3.0 | 67.5 |
| Cyclohexane | 34.0 | 98.8 | 1.2 | |
| Isooctane | 42.0 | 98.2 | 1.7 | |
| a: reaction conditions: 100 mg of 1% Ru-Co3O4,5 g of solvent,200 °C,initial pressure: 4 MPa of H2/CO2 gas (v/v=3/1), 1 h; b: calculated by considering H2 filled in the reactor as standard gas | ||||
由表2可知,溶剂对Ru-Co3O4催化剂的活性具有显著影响。水作为溶剂Ru-Co3O4表现出较低的活性,仅有8.3%;而在正丁醇中,其催化活性提高1.5倍左右,CO2转化率达到21%左右;以1,4-丁内酯和DMF为溶剂,其催化活性可进一步提高,CO2转化率分别达到26%和31%左右。一般来说,多相催化剂的加氢反应活性常常依赖于其表面活性氢的浓度。催化剂表面活性氢浓度越高,反应速率越快,同时表现出较高的活性[20]。在气相反应中,增大氢压往往有利于提高催化剂表面的活性氢浓度,进而提高催化剂的加氢活性[21];与之类似的是,在液相反应中,溶剂溶解H2的能力越高,则越有利于提高催化剂表面活性氢的浓度。因此,作者推测,Ru-Co3O4催化剂在有机溶剂中的活性比在水中的高,主要是由于反应物H2分子在有机溶剂里具有更高的溶解度所致。
因此,考察了H2分子在上述溶剂中的溶解度。为了更好地模拟真实的反应条件,在溶解度实验中,H2的压力与反应时的压力相同。由表2可知,在水中,H2的溶解度较低,为28.9 mmol/L;H2在正丁醇、1,4-丁内酯和DMF等溶剂中的溶解度是水中的1.5−3.0倍。这一结果初步证实了作者的推测,即有机溶剂高的溶氢能力提高了Ru-Co3O4的催化活性。为了进一步证实作者的猜测,选取正壬烷作为反应溶剂。发现H2在正壬烷中的溶解度也较高(表2)。催化评价结果显示,Ru-Co3O4催化剂在正壬烷中确实也表现出了较高的活性,其CO2转化率达到32%左右。当十氢萘为反应溶剂时,虽然H2在十氢萘中的溶解度低于在正壬烷中的溶解度,但催化活性却提高了50%左右,CO2转化率达到了46%左右(表2)。
为了研究溶剂作用,选取正壬烷和十氢萘作为研究对象。以D取代H,在200 °C考察了溶剂的氘代率。十氢萘和正壬烷的GC-MS分析(所用计算方法参见文献[22])结果表明,十氢萘的氘代率为4.8%,而正壬烷的氘代率只有3%。十氢萘分子结构中同时含有成环的碳原子和叔碳原子,不同碳原子上的氢在核磁共振氢谱中的化学位移相近,不易区分。为了准确确定哪个位置的碳原子的氢更易氘代,选择分别具有成环碳原子的环己烷和含有叔碳的异辛烷作为溶剂,并考察了它们的加氢反应性能。
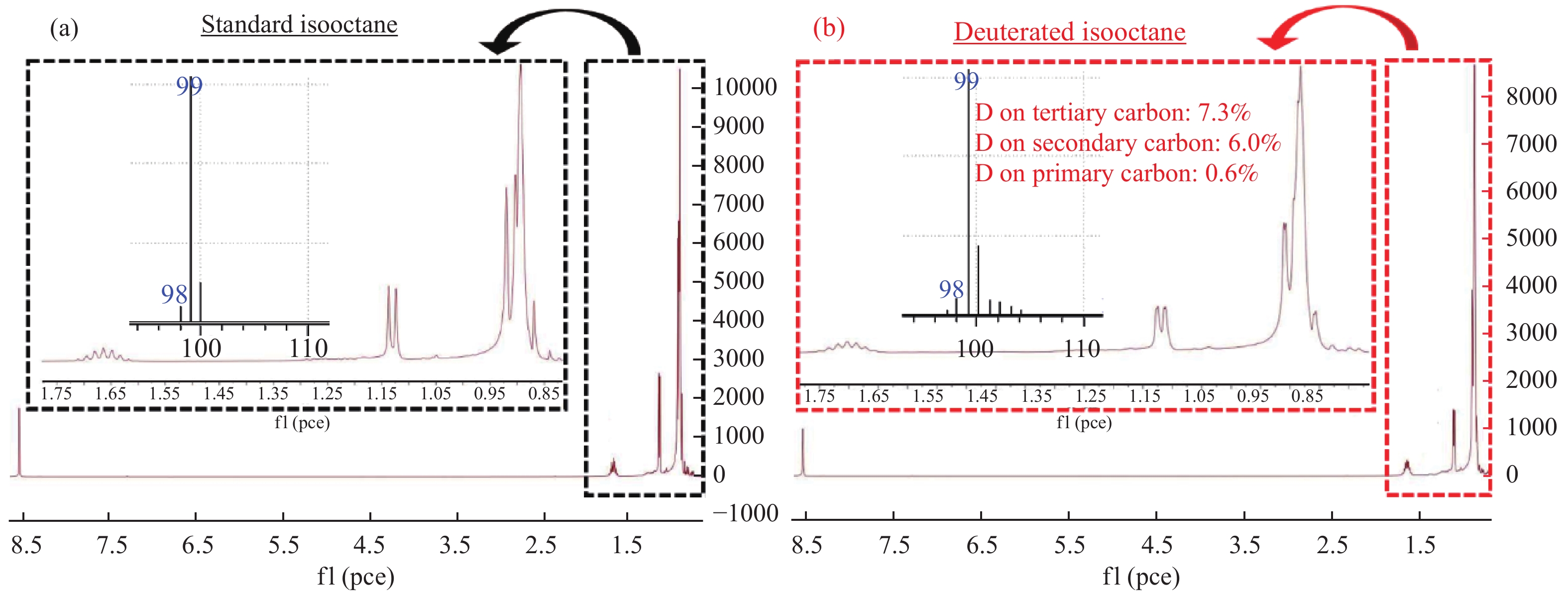
由表2可知,环己烷溶剂中CO2转化率只有34%,而在异辛烷中CO2转化率达到了42%。作者推测叔碳原子上的氢活泼性高,容易发生氘代。因此,叔碳原子可能是交换和传递活性氢的关键位点。在核磁共振氢谱中化学位移相差较大的异辛烷的氘代实验证明了这一点。图3是氘代异辛烷和未氘代异辛烷的氢核磁共振谱定量分析。可以发现,氢在叔碳上的氘代率最高,达7.3%,仲碳上的氢平均氘代率次之,为6.0%,而伯碳上的氢平均氘代率仅有0.6%。这一结果与十氢萘的氘代实验结果相吻合,说明叔碳是交换和传递活性氢的最活泼的位点。除叔碳外,仲碳也具有交换和传递活性氢的能力。
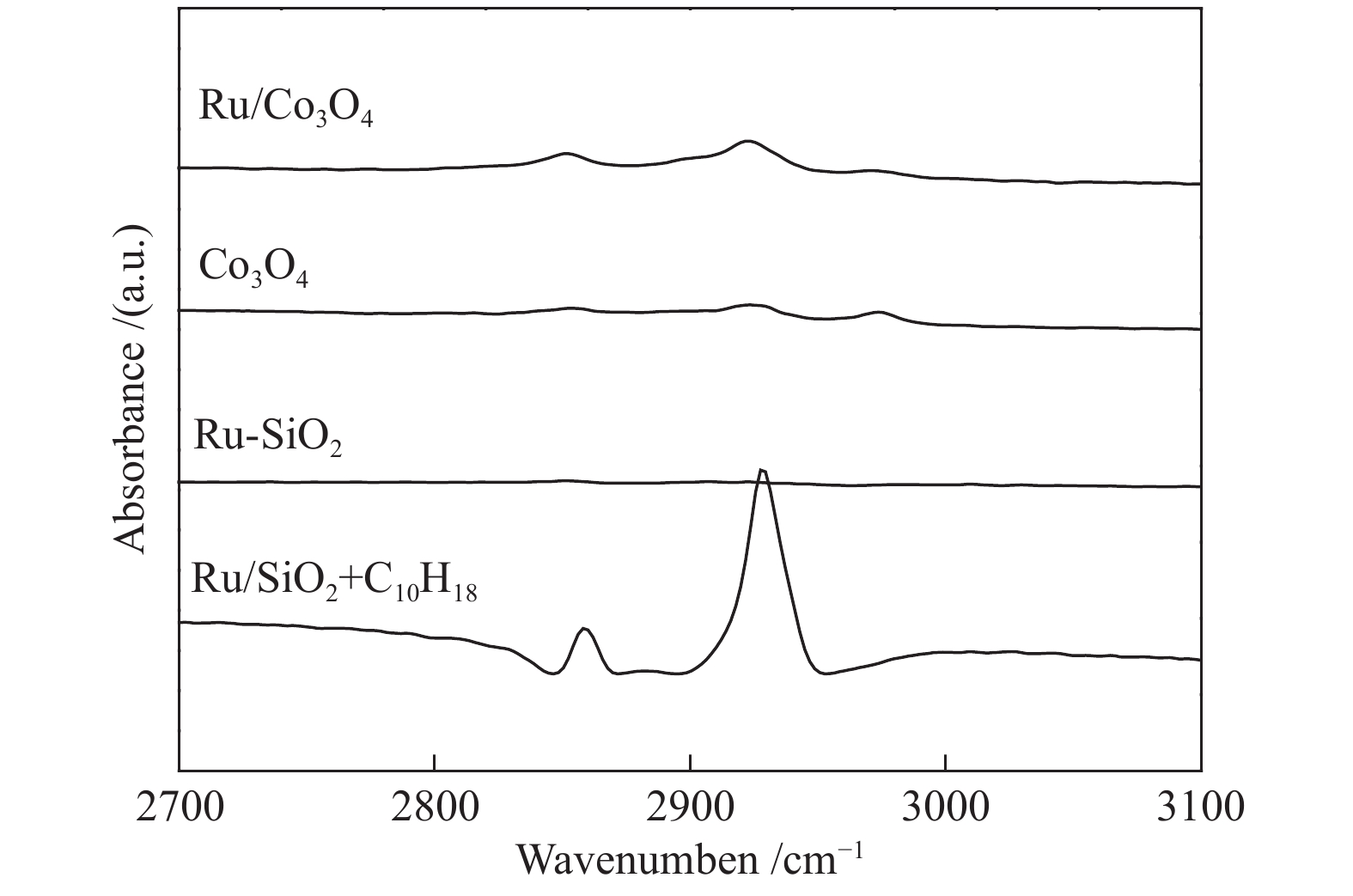
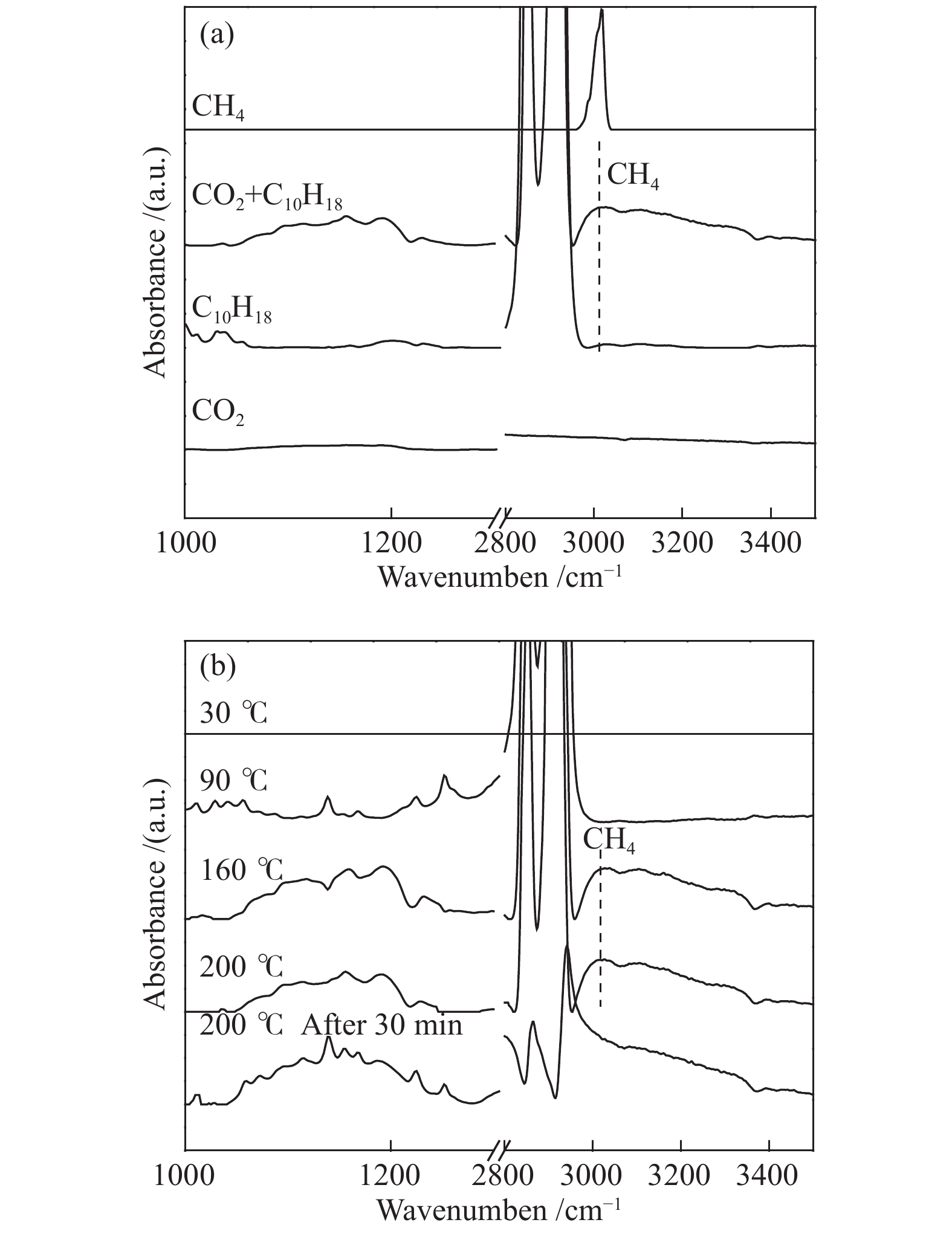
十氢萘具有较强的传递活性氢的能力(“传递氢”),那它是否可以将本身的氢作为活性氢进行加氢反应,即起到“供氢”的作用呢?这一作用要求十氢萘首先能够将氢化学吸附到催化剂表面,并活化和传递氢。图4为十氢萘在不同样品表面吸附的漫反射红外光谱。在Ru-SiO2催化剂存在时,通入十氢萘后,谱图在2858和2929 cm−1出现了振动峰,分别对应十氢萘中的−C−H和−CH2−伸缩振动征峰[23],Ar吹扫后,这些特征峰消失,说明十氢萘在Ru-SiO2表面没有发生化学吸附。然后,以相同的方式将十氢萘分别通入装有Co3O4和Ru-Co3O4的原位池中进行饱和吸附,Ar充分吹扫后谱图在2851和2922 cm−1仍然有十氢萘的−C−H和−CH2−的伸缩振动征峰;相较于Ru-SiO2上物理吸附的十氢萘,这些特征峰的峰值向低波数偏移大约7 cm−1,说明十氢萘化学吸附于Co3O4和Ru-Co3O4的表面。Co3O4上十氢萘和Ru-Co3O4催化剂上十氢萘的吸附峰位置相同,说明Ru的引入对十氢萘的化学吸附没有明显的影响。
以原位漫反射红外光谱研究了Ru-Co3O4催化剂上CO2加氢反应过程,见图5。发现在200 °C时,将催化剂分别置于CO2和十氢萘的氛围中,没有CH4的特征峰。而将催化剂置于同时含有CO2和十氢萘的氛围中,在3015 cm−1出现了甲烷峰(图5(a)),这与作者测得的甲烷标准峰位置一致,同时在1000−1300 cm−1发现含碳物种的特征峰[23],这说明了CO2与十氢萘上的氢可以反应生成CH4,十氢萘具有“供氢”的作用。进而作者又考察了,Ru-Co3O4催化剂上CO2和十氢萘在不同温度下的反应行为。如图5(b)所示,室温下没有任何红外峰,说明室温下没有反应发生;随着反应温度升高至90 °C,在1000−1300 cm−1出现红外振动峰,可以归属于吸附于催化剂表面的含碳物种。当温度进一步升高至160−200 °C时,在3015 cm−1处出现了CH4特征峰,说明CO2加氢生成CH4需要在较高温度下才能进行。值得注意的是,在200 °C反应30 min之后,气相甲烷的特征峰消失,同时十氢萘的特征峰也逐渐减弱(2800−2900 cm−1)。在30 min后,气相和物理吸附的峰消失。而离子态的十氢萘的特征峰仍存在,但十氢萘离子中没有活性氢,不能继续反应生成CH4。
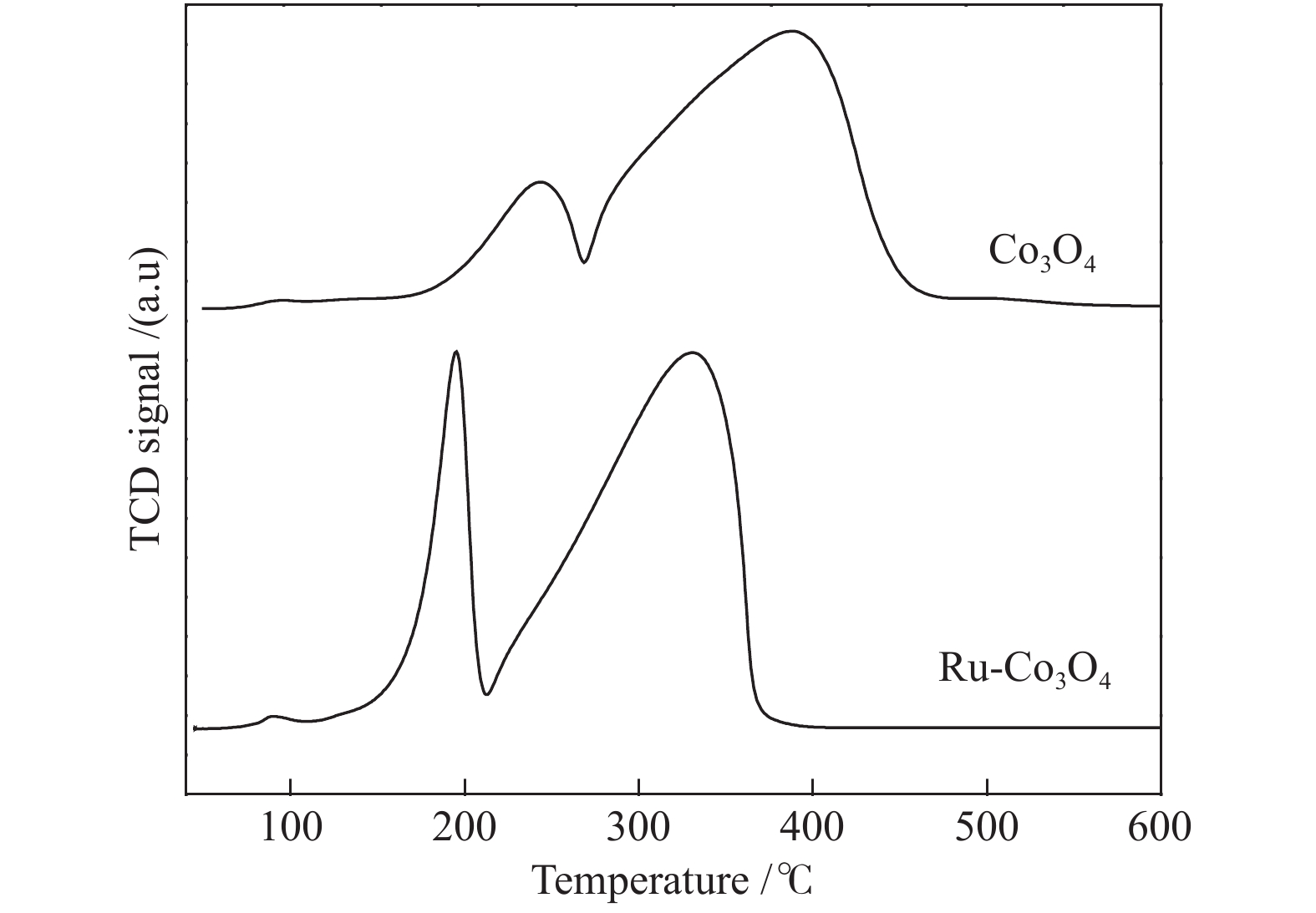
为了进一步研究反应机理,对催化剂进行了H2-TPR表征。由图6可知,Co3O4分别在242和390 °C存在两个还原峰,第一个归属于Co3O4还原为CoO的峰;第二个归属于CoO还原为Co的峰[24]。Ru的引入使Co3O4的还原温度向低温方向发生了明显的偏移,这是由于Ru具有良好的活化氢气的能力,产生的活性氢通过氢溢流促进了Co3O4的还原。
以上结果表明,十氢萘起到活性氢的载体作用,即起到“供氢”的作用。H2的活化主要有两条路线,一条是氢气吸附在Ru上活化生成活性氢并与CO2反应生成甲烷(图7(a));另一条是H2被吸附在Co3O4表面的十氢萘离子(失去活性氢之后所形成的)活化,随后与CO2反应生成CH4(图7(b))。实际上,十氢萘作为“供氢”溶剂已有报道,认为十氢萘的这种供氢作用是因为碳氢键的键能低于氢氢键的键能[25]。另外,Ru-Co3O4催化剂在300 °C还原后形成少量平均粒径约23 nm的金属Co颗粒,也会对H2的活化起到一定的作用。
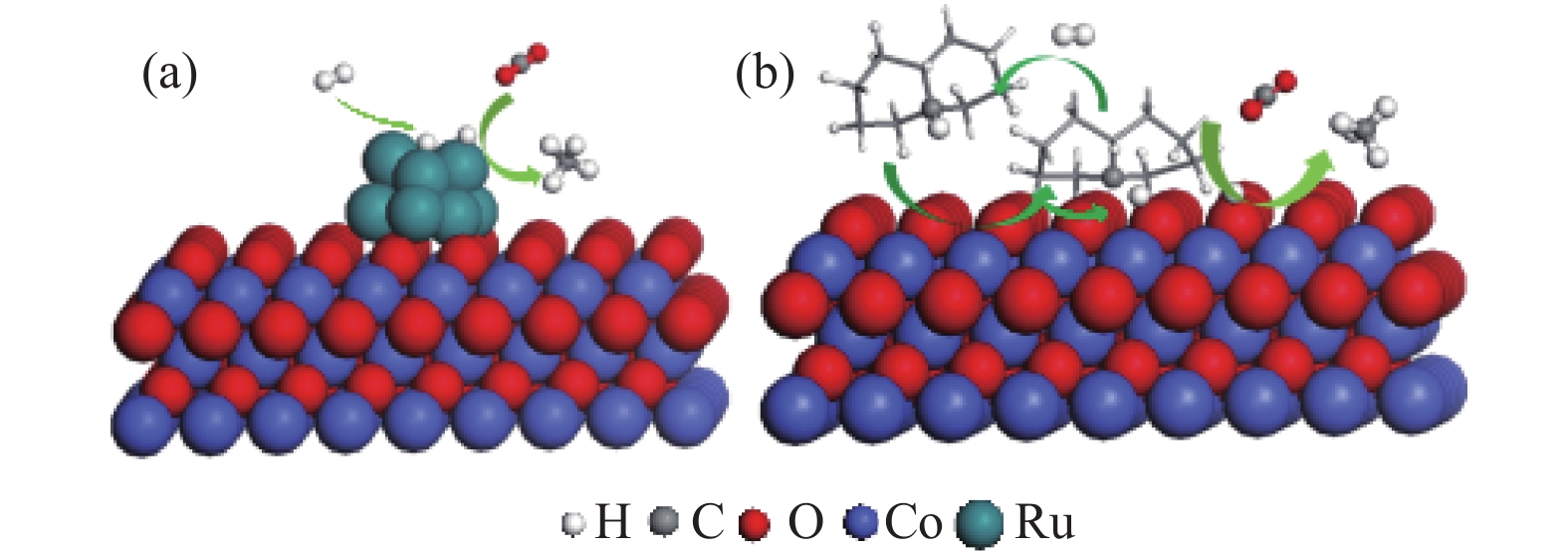
采用共沉淀法制备的Ru-Co3O4催化剂,在液相中CO2加氢制甲烷反应中显示出优良的催化性能。与传统浸渍法负载的Ru基催化剂相比,其具有高的加氢活性和甲烷选择性,以十氢萘为溶剂,在200 °C及H2/CO2 = 3∶1(v/v, 4 MPa)条件下,其CO2转化率达到45.6%,CH4的选择性达到97%左右。同位素标定实验和原位红外光谱表明,十氢萘和异辛烷对催化活性具有明显的促进作用。十氢萘首先起到交换和传递活性氢的作用,即起到“活化氢”的作用,从而有利于提高反应的活性;相比于伯碳和仲碳原子,叔碳原子上的氢活性更高,说明叔碳是交换和传递活性氢的最活泼的位点。H2在Ru-Co3O4催化剂表面活化主要有两条路线,一条是氢气吸附在Ru上活化生成活性氢并与CO2反应生成甲烷;另一条是H2被吸附在Co3O4表面的十氢萘离子活化,再与CO2反应生成CH4,两条路径同时作用使反应活性大幅提高。
DRESSELHAUS M S, THOMAS I L. Alternative energy technologies[J]. Nature,2001,414(6861):332−337. doi: 10.1038/35104599
SAKAKURA T, CHOI J-C, YASUDA H. Transformation of carbon dioxide[J]. Chem Rev,2007,107(6):2365−2387. doi: 10.1021/cr068357u
GOEPPERT A, CZAUN M, JONES J-P, PRAKASH G K S, OLAH G A. Recycling of carbon dioxide to methanol and derived products-closing the loop[J]. Chem Soc Rev,2014,43(23):7995−8048. doi: 10.1039/C4CS00122B
POROSOFF M D, YAN B, CHEN J G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities[J]. Energy Environ Sci,2016,9(1):62−73.
KONDRATENKO E V, MUL G, BALTRUSAITIS J, LARRAZABAL G O, PEREZ-RAMIREZ J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes[J]. Energy Environ Sci,2013,6(11):3112−3135.
HAVRAN V, DUDUKOVIC M P, LO C S. Conversion of methane and carbon dioxide to higher value products[J]. Ind Eng Chem Res,2011,50(12):7089−7100.
OLAJIRE A A. Recent progress on the nanoparticles-assisted greenhouse carbon dioxide conversion processes[J]. J CO2 Util,2018,24:522−547. doi: 10.1016/j.jcou.2018.02.012
BERTINI F, GLATZ M, GORGAS N, STOEGER B, PERUZZINI M, VEIROS L F, KIRCHNER K, GONSALVI L. Carbon dioxide hydrogenation catalysed by well-defined Mn(I) PNP pincer hydride complexes[J]. Chem Sci,2017,8(7):5024−5029. doi: 10.1039/C7SC00209B
GUNASEKAR G, PARK K, JEONG H, JUNG K-D, PARK K, YOON S. Molecular Rh(III) and Ir(III) catalysts immobilized on bipyridine-based covalent triazine frameworks for the hydrogenation of CO2 to formate[J]. Catalysts,2018,8(7):295. doi: 10.3390/catal8070295
SORDAKIS K, TSURUSAKI A, IGUCHI M, KAWANAMI H, HIMEDA Y, LAURENCZY G. Carbon dioxide to methanol: the aqueous catalytic way at room temperature[J]. Chem-Eur J,2016,22(44):15605−15608. doi: 10.1002/chem.201603407
DORNER R W, HARDY D R, WILLIAMS F W, DAVIS B H, WILLAUER H D. Influence of gas feed composition and pressure on the catalytic conversion of CO2 to hydrocarbons using a traditional cobalt-based Fischer-Tropsch catalyst[J]. Energy Fuels,2009,23(8):4190−4195. doi: 10.1021/ef900275m
VARGHESE J J, MUSHRIF S H. Origins of complex solvent effects on chemical reactivity and computational tools to investigate them: a review[J]. React Chem Eng,2019,4(2):165−206. doi: 10.1039/C8RE00226F
HE Z, QIAN Q, MA J, MENG Q, ZHOU H, SONG J. Water-enhanced synthesis of higher alcohols from CO2 hydrogenation over a Pt/Co3O4 catalyst under milder conditions[J]. Angew Chem Int Ed,2016,55(2):737−741. doi: 10.1002/anie.201507585
FILONENKO G A, VRIJBURG W L, HENSEN E J M, PIDKO E A. On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates[J]. J Catal,2016,343:97−105. doi: 10.1016/j.jcat.2015.10.002
CHENG S, ZENG Y, PEI Y, FAN K, QIAO M, ZONG B. Synthesis and catalysis of Pt/W-s-SBA-15 catalysts with short channel for glycerol hydrogenolysis to 1, 3-propanediol[J]. Acta Chim Sin,2019,77(10):1054−1062. doi: 10.6023/A19060219
NIE R, LIANG D, SHEN L, GAO J, CHEN P, HOU Z. Selective oxidation of glycerol with oxygen in base-free solution over MWCNTs supported PtSb alloy nanoparticles[J]. Appl Catal B: Environ,2012,127(30):212−220. doi: 10.1016/j.apcatb.2012.08.026
CHARY K V R, NARESH D, VISHWANATHAN V, SADAKANE M, UEDA W. Vapour phase hydrogenation of phenol over Pd/C catalysts: A relationship between dispersion, metal area and hydrogenation activity[J]. Catal Commun,2007,8(3):471−477. doi: 10.1016/j.catcom.2006.07.017
SALAVATI-NIASARI M, DAVAR F, MAZAHERI M, SHATERIAN M. Preparation of cobalt nanoparticles from [bis(salicylidene)cobalt(II)]–oleylamine complex by thermal decomposition[J]. J Magn Magn Mater,2008,320(3/4):575−578.
SHEN X, GARCES L-J, DING Y, LAUBERNDS K, ZERGER R P, AINDOW M, NETH E J, SUIB S L. Behavior of H2 chemisorption on Ru/TiO2 surface and its application in evaluation of Ru particle sizes compared with TEM and XRD analyses[J]. Appl Catal A: Gen,2008,335(2):187−195. doi: 10.1016/j.apcata.2007.11.017
BERTERO N M, APESTEGUÍA C R, MARCHI A J. Catalytic and kinetic study of the liquid-phase hydrogenation of acetophenone over Cu/SiO2 catalyst[J]. Appl Catal A: Gen,2008,349(1/2):100−109. doi: 10.1016/j.apcata.2008.07.014
LIU X, ZHOU W, YANG Y, CHENG K, KANG J. Design of efficient bifunctional catalysts for direct conversion of syngas into lower olefins via methanol/dimethyl ether intermediates[J]. Chem Sci,2018,9(20):4708−4718. doi: 10.1039/C8SC01597J
PRICE G L. Matrix Method for correction of mass spectra in deuterium-exchange applications[J]. Ind Eng Chem Res,1989,28(6):839−844. doi: 10.1021/ie00090a028
DAS T, DEO G. Synthesis, characterization and in situ DRIFTS during the CO2 hydrogenation reaction over supported cobalt catalysts[J]. J Mol Catal A: Chem,2011,350(1/2):75−82. doi: 10.1016/j.molcata.2011.09.008
HONG J, MARCEAU E, KHODAKOV A Y, GABEROVA L, GRIBOVAL-CONSTANT A, GIRARDON J-S, LA FONTAINE C, BRIOIS V. Speciation of ruthenium as a reduction promoter of silica-supported Co catalysts: A time-resolved in situ XAS investigation[J]. ACS Catal,2015,5(2):1273−1282. doi: 10.1021/cs501799p
BUSCA G, LORENZELLI V. Infrared spectroscopic identification of species arising from reactive adsorption of carbon oxides on metal-oxide surfaces[J]. Mater Chem,1982,7(1):89−126. doi: 10.1016/0390-6035(82)90059-1

图 1 Ru-Co3O4的SEM((a)、(b))和TEM照片(c)
Figure 1 SEM images ((a), (b)) and TEM images (c) of Ru-Co3O4

图 2 不同焙烧样品的XRD谱图
Figure 2 XRD patterns of calcined catalysts (a): Co3O4; (b): Ru-Co3O4; (c): Ru-SiO2; (d): Ru-CeO2; (e): Ru-ZrO2; (f): Ru-TiO2
1: Co3O4; 2: SiO2; 3: Ru; 4: CeO2; 5, 6: the characteristic peaks of TiO2 with different crystal forms (5 is auatase and 6 is rutile); 7: ZrO2

图 3 异辛烷标样(a)和氘代异辛烷(b)的核磁共振色谱
Figure 3 Nuclear magnetic resonance chromatograms of standard isooctane (a) and of deuterated isooctane (b)

图 4 室温下不同样品吸附十氢萘的漫反射红外吸附谱图
Figure 4 DRIFTS spectra for adsorption of decalin on the surfaces of various samples at room temperature

图 5 Ru-Co3O4催化剂表面CO2加氢的漫反射红外谱图:(a) 200 °C; (b) 不同温度下
Figure 5 DRIFTS spectra of CO2 hydrogenation to CH4 at 200 oC on Ru-Co3O4 in CO2, decalin, (CO2+decalin), and DRIFTS spectrum of standard CH4 (from the bottom to the top direction) (a); and DRIFTS spectra of CO2 hydrogenation to CH4 at 200 oC on Ru-Co3O4 in (CO2+decalin) at different reaction temperatures (b)

图 7 十氢萘溶剂中Ru-Co3O4表面CO2加氢生成甲烷的反应路径
Figure 7 Schematic presentation for catalytic mechanism of CO2 hydrogenation to CH4 over Ru-Co3O4 in decalin solvent
表 1 不同Ru-基催化剂液相CO2加氢制甲烷的催化性能
Table 1. Catalytic results of various supported Ru catalysts for liquid-phase hydrogenation of CO2 to CH4
| Entry | Catalyst | CO2 conversion /% |
Selectivity of
hydrocarbons/mol% |
|||
| CH4 | C2−5 | CO | ||||
| 1 | Ru/SiO2 | 2.3 | 97.1 | 2.9 | 0 | |
| 2 | Ru/CeO2 | 5.4 | 98.8 | 1.2 | 0 | |
| 3 | Ru/ZrO2 | 11.3 | 97.9 | 2.1 | 0 | |
| 4 | Ru/TiO2 | 15.2 | 97.1 | 2.9 | 0 | |
| 5 | Ru-Co3O4 | 45.6 | 97.0 | 2.9 | 0.1 | |
| 6 | Co3O4 | 34.0 | 97.7 | 2.2 | 0.1 | |
| reaction conditions: 100 mg of catalyst, 5 g decalin, 200 °C, initial pressure: 4 MPa of H2/CO2 gas (v/v = 3∶1), 1 h | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 不同溶剂中Ru-Co3O4催化剂上CO2液相加氢制甲烷性能的影响a
Table 2. Catalytic results of Ru-Co3O4 for liquid-phase hydrogenation of CO2 to CH4 in different solventsa
| Solvent | CO2 conversion/% | Product selectivity/% | Solubility b/(mmol·L−1) | |
| CH4 | C2−5 | |||
| Water | 8.3 | 88.7 | 11.0 | 28.9 |
| Butyl alcohol | 21.4 | 88.7 | 11.1 | 87.2 |
| 1,4- butyrolactone | 26.2 | 95.7 | 4.2 | 42.6 |
| DMF | 31.0 | 89.4 | 10.4 | 43.2 |
| n-Nonane | 31.9 | 98.5 | 1.5 | 93.5 |
| Decalin | 45.6 | 97.0 | 3.0 | 67.5 |
| Cyclohexane | 34.0 | 98.8 | 1.2 | |
| Isooctane | 42.0 | 98.2 | 1.7 | |
| a: reaction conditions: 100 mg of 1% Ru-Co3O4,5 g of solvent,200 °C,initial pressure: 4 MPa of H2/CO2 gas (v/v=3/1), 1 h; b: calculated by considering H2 filled in the reactor as standard gas | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们