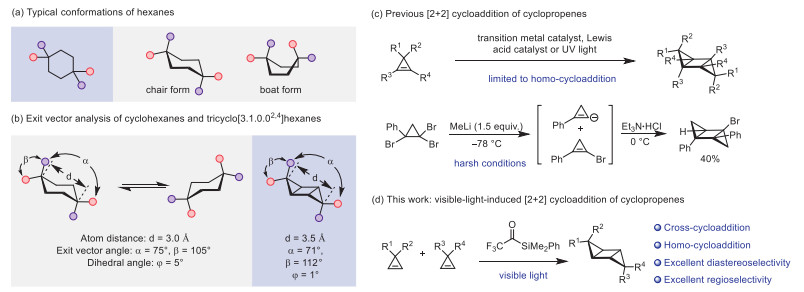
Scheme 1.
Background and our strategy for the synthesis of rigidified cyclohexanes by visible-light-induced photosensitized [2 + 2] cycloaddition of cyclopropenes.

Cyclohexanes are important structural motifs in pharmaceutical chemistry and organic synthesis [1–4]. In the dynamic equilibrium of a series of conformations, chair form and boat form are two typical conformations that can interconvert to each other (Scheme 1a). Moreover, the chair cyclohexanes also have conformational isomers (Scheme 1b). However, the inherent flexibility of cyclohexanes negatively affects their binding selectivity and affinity and their metabolic stability, thereby limiting the use of cyclohexane derivatives as medicines. Imposing conformational constraints on lead compounds provides an important strategy for structural modification and optimization [5]. Recently, Brown, Fessard and co-workers reported an elegant application of 2,6-disubstituted [2]-ladderanes as rigidified 1,3-disubstituted cyclohexanes [6]. We envisioned that tricyclo[3.1.0.02,4]hexanes can be treated as a kind of rigidified 3,3,6,6-substituted cyclohexanes, since their geometric properties are similar (Scheme 1b, for details of the DFT calculation of the conformations of selected cyclohexanes and tricyclo[3.1.0.02,4]hexanes, see Supporting information).
However, traditional methods for the synthesis of tricyclo[3.1.0.02,4]hexanes relied on homo-dimerization of cyclopropenes promoted by transition-metal catalyst (e.g., Pd, Rh and Ni complexes) and Lewis acids (Scheme 1c) [7–21]. Cycloaddition reaction of cyclopropenes under UV irradiation features simpler conditions, but they are still limited to homo-dimerization, probably because both cyclopropenes could be excited by the high-energy light (Scheme 1c) [22–32]. The only example of formal cross-cycloaddition between different cyclopropenes employed a stoichiometric amount of methyllithium reagent at low temperature, but the cyclopropene intermediates need to be in situ generated from the same tribrominated cyclopropane, and only 40% yield was obtained (Scheme 1c) [21]. Under this context, the development of practical [2 + 2] cycloaddition of cyclopropenes, especially cross [2 + 2] cycloaddition reaction under mild conditions, is highly desired. In recent years, visible-light-induced triplet-triplet energy transfer catalysis has emerged as a milder strategy to sensitize the substrates for efficient transformations [33–49]. Recently, we found that the triplet-triplet energy transfer between acylsilanes and Eosin Y could protect the acylsilanes from light-induced decomposition, indicating the potential of acylsilanes as energy transfer catalysts [47]. Herein, we report the first application of trifluoroacetylsilanes as photosensitizers in the visible-light-induced cycloaddition of cyclopropenes (Scheme 1d). The reactions show excellent chemoselectivity and diastereoselectivity for the cross-cycloaddition reactions. A preliminary mechanistic study indicates aggregation promoted sensitization of cyclopropenes by the trifluoroacetylsilane.
At the outset, we investigated the conditions of cross-cycloaddition reaction with cyclopropenes 1a and 2a as the model substrates (Table 1). Through careful screening of the reaction parameters including photosensitizers, solvents, concentrations, light sources, ratio of reactants and reaction time (for details, see Supporting information), we found that when trifluoroacetylsilane 3a (1.0 equiv.) was used as a photosensitizer, the reaction of 1a and 2a (2.0 equiv.) in DCE (0.4 mol/L) at room temperature under 450 nm LEDs (6 W) for 36 h afforded cross-cycloaddition product 4a in 70% yield with 95:5 dr (74% isolated yield, Table 1, entry 1), accompanied by the formation of homo-cycloaddition product 5a in 30% yield with 74:26 dr and homo-cycloaddition product 6a in 26% yield. There was 24% conversion of acylsilane 3a observed (Table 1, entry 1). The reaction could be performed with a catalytic amount of 3a (5 mol%), but the efficiency is greatly reduced, and only 21% conversion of cyclopropene 1a and 12% yield of 4a were obtained (Table 1, entry 2). With 2 mol% of fac-Ir(ppy)3, no desired cycloaddition product was obtained, while 7% conversion of 1a was found (Table 1, entry 3). When benzophenone was used instead of trifluoroacetylsilane 3a under 400 nm LEDs (6 W), no conversion of substrate 1a was detected (Table 1, entry 4). The non-fluorinated acylsilane 3b was found to be much less efficient than 3a (5% yield, Table 1, entry 5). Further investigation revealed that the solvent could significantly influence the formation of different products (Table 1, entries 6-8). When toluene was used instead of DCE, the yield of 4a decreased to 57%, and the homocoupling products 5a and 6a were obtained at 46% and 40% yield, respectively (Table 1, entry 6). When EtOAc was used as a solvent, the dr of 4a decreased to 89:11, while a synthetically useful yield was obtained (65% yield, Table 1, entry 7). With MeCN as the solvent, both the efficiency of the reaction and the diastereoselectivity for the formation of 4a decreased significantly (7% yield, 86:14 dr, Table 1, entry 8). The use of 420 nm LEDs led to only 37% conversion of 1a and 14% yield of 4a (Table 1, entry 9), probably because of the decomposition of 3a under the high-energy light irradiation. When the amount of 2a was reduced to 1.5 equiv., the reaction became slower, and the yield of 4a decreased to 24% (Table 1, entry 10). Control experiments showed that the reaction does not proceed in the absence of trifluoroacetylsilane 3a or light (Table 1, entries 11 and 12).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||||||
| Entry | Variation | Conversion of 1a (%) | 4a | 5a | Yield of 6a (%) | |||
| Yield (%) | dr | Yield (%) | dr | |||||
| 1 | None | 100 | 70 (74)b,c | 95:5 | 30 | 74:26 | 26 | |
| 2 | 5 mol% of 3a instead of 1.0 equiv. of 3a | 21 | 12 | – | 5 | – | 2 | |
| 3 | 2 mol% of fac-Ir(ppy)3 instead of 3a | 7 | 0 | – | 0 | – | 0 | |
| 4 | 1.0 equiv. of benzophenone (400 nm LEDs) instead of 3a | 0 | 0 | – | 0 | – | 0 | |
| 5 | 1.0 equiv. of 3b (420 nm LEDs) instead of 3a | 17 | 5 | – | 4 | – | 3 | |
| 6 | Toluene instead of DCE | 100 | 57 | 95:5 | 46 | 84:16 | 40 | |
| 7 | EtOAc instead of DCE | 100 | 65 | 89:11 | 35 | 76:24 | 66 | |
| 8 | MeCN instead of DCE | 10 | 7 | 86:14 | 2 | – | 5 | |
| 9 | 420 nm instead of 450 nm LEDs | 37 | 14d | – | 8 | – | 2 | |
| 10 | 1.5 equiv. of 2a | 43 | 24 | 95:5 | 19 | 72:28 | 3 | |
| 11 | No 3a | 0 | 0 | – | 0 | – | 0 | |
| 12 | No light | 0 | 0 | – | 0 | – | 0 | |
| a Unless otherwise noted, the reaction conditions were as follows: 1a (0.10 mmol), 2a (0.20 mmol), 3a (0.10 mmol), DCE (0.40 mol/L), irradiation by 450 nm LEDs under Ar at 26 ℃ for 36 h; the conversion of 1a, the yield of 4a, 5a and 6a, the dr of 4a and 5a were determined by 1H NMR spectroscopy with BrCH2CH2Br as an internal standard. DCE: 1,2-dichloroethane. b Isolated yield. c 24% conversion of 3a was observed by 19F NMR with PhCF3 as an internal standard. d 100% conversion of 3a was observed by 19F NMR with PhCF3 as an internal standard. |
||||||||
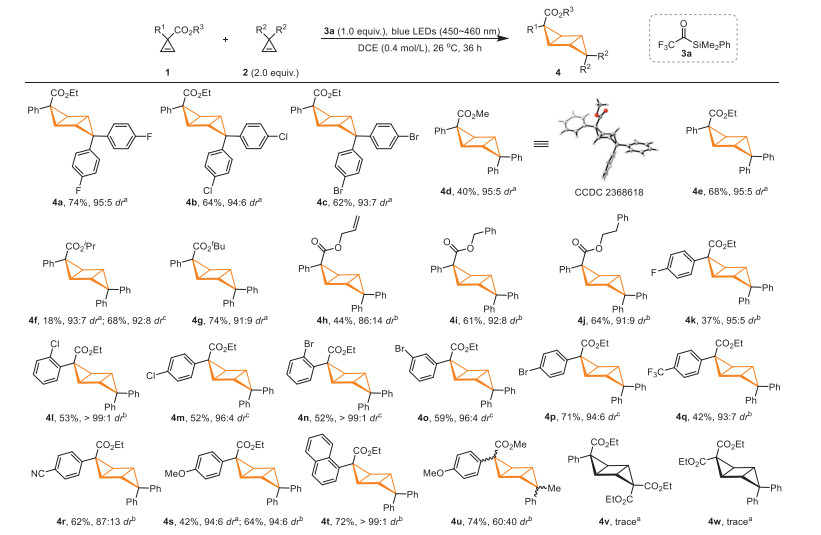
With the optimized conditions in hand, we examined the substrate scope of cyclopropenes in [2 + 2] cross-cycloaddition reactions (Scheme 2). Diarylcyclopropenes with F, Cl or Br substituents on the aromatic rings performed well, providing the desired products 4a-4c in 62%-74% yield with 93:7-95:5 dr. It was further found that various ester groups could be tolerated by the reaction (4d-4j, 40%-74% yield), and the cross-cycloaddition product was obtained in higher diastereoselectivity with the cyclopropene that contain less sterically hindered ester group (4d, 95:5 dr vs. 4g, 91:9 dr). Compound 4d was obtained in 40% yield with 95:5 dr, and the relative configuration of its major isomer was determined by X-ray crystal structure analysis (CCDC: 2368618). The relative configurations of the major isomers of other products were assigned accordingly. It is worth noting that an aliphatic alkene can be tolerated by this reaction, and compound 4h was obtained in 44% yield with 86:14 dr. Furthermore, various aryl ethoxycarbonyl cyclopropenes have been studied with diphenylcyclopropene 2d as the reaction partner, and we found that a variety of functional groups such as F, Cl, Br, CF3, CN and OMe could be tolerated, affording compounds 4k-4s in 37%-71% yield with 87:13~ > 99:1 dr. A naphthalene-containing product 4t was also successfully prepared in 72% yield with 99:1 dr, although increasing the loading of sensitizer 3a to 2 equiv. was needed to improve the reaction rate. The reaction can also proceed when (1-methylcycloprop-2-en-1-yl)benzene 2e was used as the reaction partner, and the product 4u was obtained in 74% yield with 60:40 dr. However, compounds 4v and 4w were not detected, when diethyl cycloprop-2-ene-1,1-dicarboxylate was used as the substrate.
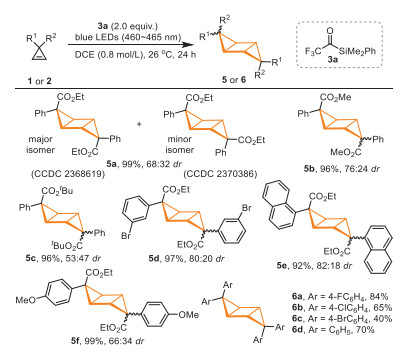
Subsequently, we investigated the scope of cyclopropenes in the visible-light-induced photosensitized [2 + 2] homo-cycloaddition reactions (Scheme 3). The investigation of the ester group substituents on cyclopropenes showed that the size of the ester group did not affect the yield of the product, but affected the diastereoselectivity of the reaction (5a-5c, 96%-99% yield, 53:47-76:24 dr). The configurations of the two diastereoisomers of compound 5a were determined by X-ray crystal structure analysis (major isomer: CCDC 2368619; minor isomer: CCDC 2370386). Cyclopropenes containing Br or OMe substituted aromatic rings and naphthyl ring were also suitable substrates, affording corresponding homo-cycloaddition products 5d-5f in 92%-99% yield with 66:34-82:18 dr. A wide range of diarylcyclopropenes were also suitable substrates for the homo-cycloaddition reaction, providing products 6a-6d in 40%-84% yield.
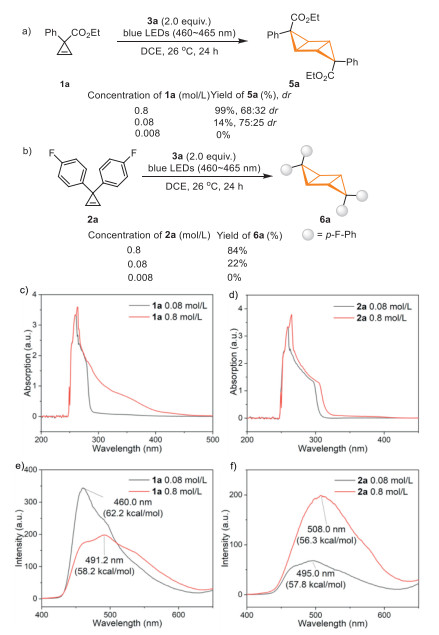
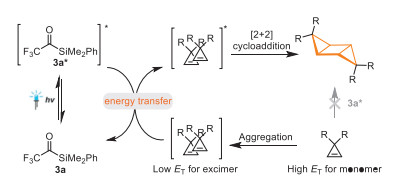
Acetylsilanes are known to be able to generate carbenes [50–71]. The unique application of trifluoroacetylsilane 3a as a photocatalyst in mediating visible-light-induced [2 + 2] cycloaddition reaction promoted us to carry out the preliminary mechanistic study. The DFT calculation revealed that trifluoroacetylsilane 3a possesses lower triplet energy (ET = 56.7 kcal/mol in DCE) than those of 1a and 2a (1a, ET = 64.8 kcal/mol in DCE; 2a, ET = 61.1 kcal/mol in DCE), which might disfavor the triplet-triplet energy transfer mechanism (for details of the DFT calculations, see Supporting information). However, Wu and co-workers reported an aggregation-induced photocatalytic [2 + 2] cycloaddition of two terminal alkenes [72]. They revealed that aryl terminal alkenes tend to aggregate to form excimers or exciplexes that possess lower energies than their monomeric forms in their triplet excited states, thereby, energy transfer from excited photocatalyst to supramolecular self-assembly is feasible. To test whether this aggregation effect exists in our reaction, we performed further experimental, spectral and computational studies (Fig. 1). Firstly, we found that higher concentration is beneficial for the reaction (Figs. 1a and b). Secondly, the UV–vis absorption spectra of cyclopropenes 1a and 2a showed significant redshift when the concentration increased, indicating the assembly of cyclopropenes to generate species with lower excited state energy (Figs. 1c and d). In order to further probe this interaction, the phosphorescence spectra of the substrates 1a and 2a at 77 K were measured (Figs. 1e and f). When the concentration of 1a increased from 0.08 mol/L to 0.8 mol/L, the phosphorescence peak at 460 nm was replaced by the new signals of excimer at 491.2 nm, suggesting a decreased triplet energy (ET(460 nm) = 62.2 kcal/mol to ET(491 nm) = 58.2 kcal/mol). When the concentration of substrate 2a increased from 0.08 mol/L to 0.8 mol/L, the phosphorescence peak at 495 nm was replaced by new signals of excimer at 508 nm that also suggested decreased triplet energy (ET(495 nm) = 57.8 kcal/mol to ET(508 nm) = 56.3 kcal/mol). Therefore, we believe that the aggregation effect of cyclopropenes in solution reduced their excited state energy, and thus promoted the visible-light-induced [2 + 2] homocoupling and cross-coupling of cyclopropenes. Therefore, we proposed a possible reaction pathway in Fig. 2. Under light irradiation, trifluoroacetylsilane 3a was transformed to the excited state, which subsequently sensitized the aggregated cyclopropenes, facilitating the [2 + 2] cycloaddition.
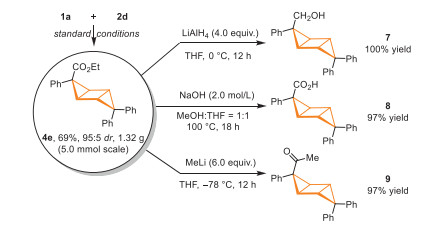
The practicality of the protocol was demonstrated by a gram-scale reaction between cyclopropene 1a and 2d and the yield and diastereoselectivity were not significantly affected by the increased reaction scale, paving a practical route for large-scale preparation of tricyclo[3.1.0.02,4]hexanes. In order to show the synthetic potential of this method, we have performed several down-stream transformations of the cross-cycloaddition product 4e (Scheme 4). The major isomer of compound 4e was isolated in pure form and subjected to a reduction reaction with LiAlH4 at 0 ℃, and alcohol 7 was isolated in 100% yield. Under the treatment with NaOH in MeOH/THF at 100 ℃ for 18 h, the ester group of 4e was hydrolyzed, and carboxylic acid 8 was obtained in 97% yield. The ester group of 4e could also be converted to ketone by treating 4e with MeLi at −78 ℃, affording compound 9 in 97% yield.
In conclusion, we have developed a photosensitized [2 + 2] cycloaddition of cyclopropenes to synthesize substituted tricyclo[3.1.0.02,4]hexanes as rigidified cyclohexanes. Excellent chemoselectivity and diastereoselectivity have been achieved in the cross-cycloaddition reactions. A trifluoroacetylsilane was found to be a unique photosensitizer. The mechanism study supports that the aggregation effect of cyclopropenes in solution promotes the energy transfer from excited trifluoroacetylsilane to aggregated cyclopropenes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Meixin Wang: Investigation. Yizhi Zhang: Investigation. Shanshan Liu: Investigation. Xiao Shen: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
We are grateful to National Key R & D Program of China (No. 2022YFA1506100), National Natural Science Foundation of China (Nos. 22471201, 21901191), Fundamental Research Funds for the Central Universities (No. 2042023kf0202) and Wuhan University for financial support. We thank Ran Zhang from the core Facility of Wuhan University for the assistance with X-Ray structure analysis. The theoretical calculations were performed on the supercomputing system in the Supercomputing Center of Wuhan University.
Supplementary material associated with this article can be found, in the online version, at doi:
C.M. Marson, Chem. Soc. Rev. 40 (2011) 5514–5533. doi: 10.1039/c1cs15119c
X. Jiang, R. Wang, Chem. Rev. 113 (2013) 5515–5546. doi: 10.1021/cr300436a
E.C. Cherney, L. Zhang, S. Nara, et al., ACS Med. Chem. Lett. 12 (2021) 288–294. doi: 10.1021/acsmedchemlett.0c00668
N. Moss, Z. Xiong, M. Burke, et al., Bioorg. Med. Chem. Lett. 22 (2012) 7189–7193.
Z. Fang, Y. Song, P. Zhan, Q. Zhang, X. Liu, Future Med. Chem. 6 (2014) 885–901. doi: 10.4155/fmc.14.50
R.C. Epplin, S. Paul, L. Herter, et al., Nat. Commun. 13 (2022) 6056.
R.L. Baird, F.J. Weigert, J.R. Shapley, J. Am. Chem. Soc. 92 (1970) 6630–6635. doi: 10.1021/ja00725a041
P. Binger, A. Brinkmann, Chem. Ber. 111 (1978) 2689–2695. doi: 10.1002/cber.19781110723
P. Binger, J. McMeeking, U. Schuchardt, Chem. Ber. 113 (1980) 2372–2382. doi: 10.1002/cber.19801130706
P. Binger, U. Schuchardt, Chem. Ber. 114 (1981) 1649–1655. doi: 10.1002/cber.19811140508
P. Binger, B. Biedenbach, Chem. Ber. 120 (1987) 601–605. doi: 10.1002/cber.19871200422
P. Binger, H. Schäfer, Tetrahedron Lett. 16 (1975) 4673–4676.
T.L. Arrowood, S.R. Kass, Tetrahedron 55 (1999) 6739–6748.
M.A. Smith, H.G. Richey, Organometallics 26 (2007) 609–616. doi: 10.1021/om060766t
S. Kishida, M. Takano, T. Sekiya, Y. Ukaji, K. Endo, J. Org. Chem. 87 (2022) 14833–14839. doi: 10.1021/acs.joc.2c01261
K.B. Wiberg, G. Bonneville, Tetrahedron Lett. 23 (1982) 5385–5388.
W.E. Billups, L.J. Lin, Tetrahedron 42 (1986) 1575–1579.
K.B. Wiberg, D.R. Artis, G. Bonneville, J. Am. Chem. Soc. 113 (1991) 7969– 7979. doi: 10.1021/ja00021a024
A.R. Al Dulayymi, M.S. Baird, Tetrahedron 52 (1996) 10955–10968.
G.A. Lee, C.Y. Chang, J. Org. Chem. 69 (2004) 8949–8951. doi: 10.1021/jo048889f
G.A. Lee, W.C. Wang, S.F. Jiang, C.Y. Chang, R.T. Tsai, J. Org. Chem. 74 (2009) 7994–7997. doi: 10.1021/jo901636k
H.H. Stechl, Chem. Ber. 97 (1964) 2681–2688. doi: 10.1002/cber.19640970936
N. Obata, I. Moritani, Tetrahedron Lett. 7 (1966) 1503–1508.
N. Obata, I. Moritani, Bull. Chem. Soc. Jpn. 39 (1966) 2250–2255. doi: 10.1246/bcsj.39.2250
C. Deboer, R. Breslow, Tetrahedron Lett. 8 (1967) 1033–1038.
H. Dürr, Justus Liebigs Ann. Chem. 723 (1969) 102–110. doi: 10.1002/jlac.19697230112
C.D. DeBoer, D.H. Wadsworth, W.C. Perkins, J. Am. Chem. Soc. 95 (1973) 861–869. doi: 10.1021/ja00784a039
W. Eberbach, Chem. Ber. 107 (1974) 3287–3325. doi: 10.1002/cber.19741071016
J.A. Pincock, A.A. Moutsokapas, Can. J. Chem. 55 (1977) 979–985. doi: 10.1139/v77-137
A. Padwa, Acc. Chem. Res. 12 (1979) 310–317. doi: 10.1021/ar50141a002
A. Padwa, G.D. Kennedy, G.R. Newkome, F.R. Fronczek, J. Am. Chem. Soc. 105 (1983) 137–139. doi: 10.1021/ja00339a036
E.I. Klimova, M.M. Garcia, T. Klimova, C.A. Toledano, R.A. Toscano, J. Organomet. Chem. 605 (2000) 89–95.
F. Strieth-Kalthoff, M.J. James, M. Teders, L. Pitzer, F. Glorius, Chem. Soc. Rev. 47 (2018) 7190–7202. doi: 10.1039/c8cs00054a
Q.Q. Zhou, Y.Q. Zou, L.Q. Lu, W.J. Xiao, Angew. Chem. Int. Ed. 58 (2019) 1586–1604. doi: 10.1002/anie.201803102
T. Neveselý, M. Wienhold, J.J. Molloy, R. Gilmour, Chem. Rev. 122 (2022) 2650–2694. doi: 10.1021/acs.chemrev.1c00324
P. Rana, N. Singh, P. Majumdar, S.P. Singh, Coordin. Chem. Rev. 470 (2022) 214698.
R. Hojo, A.M. Polgar, Z.M. Hudson, ACS Sustain. Chem. Eng. 10 (2022) 9665–9678. doi: 10.1021/acssuschemeng.2c01426
J. Molloy, M. Schäfer, M. Wienhold, et al., Science 369 (2020) 302–306. doi: 10.1126/science.abb7235
K.M. Nakafuku, Z. Zhang, E.A. Wappes, et al., Nat. Chem. 12 (2020) 697–704. doi: 10.1038/s41557-020-0482-8
M.R. Becker, E.R. Wearing, C.S. Schindler, Nat. Chem. 12 (2020) 898–905. doi: 10.1038/s41557-020-0541-1
Q. Cheng, J. Chen, S. Lin, T. Ritter, J. Am. Chem. Soc. 142 (2020) 17287–17293. doi: 10.1021/jacs.0c08248
F.M. Hormann, C. Kerzig, T.S. Chung, et al., Angew. Chem. Int. Ed. 59 (2020) 9659–9668. doi: 10.1002/anie.202001634
L. Tian, N.A. Till, B. Kudisch, D.W.C. MacMillan, G.D. Scholes, J. Am. Chem. Soc. 142 (2020) 4555–4559. doi: 10.1021/jacs.9b12835
M. Zhu, X.L. Huang, H. Xu, et al., CCS Chem. 3 (2021) 652–664. doi: 10.31635/ccschem.020.202000254
J. Ma, S. Chen, P. Bellotti, et al., Science 371 (2021) 1338–1345. doi: 10.1126/science.abg0720
Y. Liu, D. Ni, B.G. Stevenson, et al., Angew. Chem. Int. Ed. 61 (2022) e20220072.
Y. Zhang, Y. Zhang, C. Ye, et al., Nat. Commun. 13 (2022) 6111.
J. Wang, Q. Fu, S. Cao, et al., J. Am. Chem. Soc. 146 (2024) 22840–22849. doi: 10.1021/jacs.4c08290
Y. Du, L. Yao, X. Li, Y. Guo, Chin. Chem. Lett. 34 (2023) 107512.
G. Zhou, Z. Guo, X. Shen, Angew. Chem. Int. Ed. 62 (2023) e202217189.
D.L. Priebbenow, Adv. Synth. Catal. 362 (2020) 1927–1946. doi: 10.1002/adsc.202000279
H.J. Zhang, D.L. Priebbenow, C. Bolm, Chem. Soc. Rev. 42 (2013) 8540–8571. doi: 10.1039/c3cs60185d
Y. Guo, G. Zhou, X. Shen, Chin. J. Chem. 42 (2024) 887–902. doi: 10.1002/cjoc.202300569
A.G. Brook, J.M. Duff, J. Am. Chem. Soc. 89 (1967) 454–455. doi: 10.1021/ja00978a053
R.A. Bourque, P.D. Davis, J.C. Dalton, J. Am. Chem. Soc. 103 (1981) 697–699. doi: 10.1021/ja00393a048
R.F. Cunico, A.R. Motta, Org. Lett. 7 (2005) 771–774. doi: 10.1021/ol040066p
Z. Shen, V.M. Dong, Angew. Chem. Int. Ed. 48 (2009) 784–786. doi: 10.1002/anie.200804854
K. Ito, H. Tamashima, N. Iwasawa, H. Kusama, J. Am. Chem. Soc. 133 (2011) 3716–3719. doi: 10.1021/ja1102597
P. Becker, D.L. Priebbenow, R. Pirwerdjan, C. Bolm, Angew. Chem. Int. Ed. 53 (2014) 269–271. doi: 10.1002/anie.201307446
J.H. Ye, L. Quach, T. Paulisch, F. Glorius, J. Am. Chem. Soc. 141 (2019) 16227–16231. doi: 10.1021/jacs.9b08960
B. Huang, M. Wei, E. Vargo, et al., J. Am. Chem. Soc. 143 (2021) 17920–17925. doi: 10.1021/jacs.1c06836
C. Stuckhardt, M. Wissing, A. Studer, Angew. Chem. Int. Ed. 60 (2021) 18605–18611. doi: 10.1002/anie.202101689
S. Sakurai, T. Inagaki, T. Kodama, M. Yamanaka, M. Tobisu, J. Am. Chem. Soc. 144 (2022) 1099–1105. doi: 10.1021/jacs.1c11497
Z. Zhu, W. Zhang, Y. Zhang, S. Liu, X. Shen, CCS Chem. 5 (2023) 325–333.
Y. Ueda, Y. Masuda, T. Iwai, et al., J. Am. Chem. Soc. 144 (2022) 2218–2224. doi: 10.1021/jacs.1c11526
A. Bunyamin, C. Hua, A. Polyzos, D.L. Priebbenow, Chem. Sci. 13 (2022) 3273–3280. doi: 10.1039/d2sc00203e
T. Inagaki, T. Kodama, M. Tobisu, Nat. Catal. 7 (2024) 132–138. doi: 10.1038/s41929-023-01081-5
Y. Liu, Z. Zhu, Y. Zhang, et al., Org. Lett. 26 (2024) 5911–5916. doi: 10.1021/acs.orglett.4c01782
G. Zhou, X. Shen, Angew. Chem. Int. Ed. 61 (2022) e202115334.
Y. Zhang, G. Zhou, X. Gong, et al., Angew. Chem. Int. Ed. 61 (2022) e202202175.
G. Zhou, Z. Guo, S. Liu, X. Shen, J. Am. Chem. Soc. 146 (2024) 4026–4035. doi: 10.1021/jacs.3c12150
Z. Liu, C. Zhou, T. Lei, et al., CCS Chem. 2 (2020) 582–588.

Scheme 1 Background and our strategy for the synthesis of rigidified cyclohexanes by visible-light-induced photosensitized [2 + 2] cycloaddition of cyclopropenes.

Scheme 2 a Reaction conditions: 1 (0.1 mmol), 2 (0.2 mmol, 2 equiv.), 3a (0.1 mmol, 1 equiv.), DCE (0.25 mL), 26 ℃, 36 h; the isolated yield is given; dr was determined by 1H NMR analysis of the unpurified reaction mixture. b 3a (0.2 mmol, 2 equiv.), 48 h. c 3a (0.3 mmol, 3 equiv.), 48 h. The configuration of the major isomer of compound 4 is shown in Table 1.

Scheme 3 Reaction conditions: 1 or 2 (0.2 mmol), 3a (0.20 mmol, 2.0 equiv.), DCE (0.25 mL), 26 ℃, 24 h; the yield refers to the isolated total yield of both diastereoisomers; dr was determined by 1H NMR analysis of the unpurified reaction mixture.

Figure 1 Mechanistic studies. (a) Concentration influence on the homo-cycloaddition of 1a. (b) Concentration influence on the homo-cycloaddition of 2a. (c) UV–vis absorption spectra of 1a in DCE at 0.08 and 0.8 mol/L. (d) UV–vis absorption spectra of 2a in DCE at 0.08 and 0.8 mol/L. (e) Phosphorescence spectra of 1a in DCE at 0.08 and 0.8 mol/L, respectively, at 77 K. (f) Phosphorescence spectra of 2a in DCE at 0.08 and 0.8 mol/L, respectively, at 77 K.
Table 1. Optimization of reaction conditions.
|
||||||||
| Entry | Variation | Conversion of 1a (%) | 4a | 5a | Yield of 6a (%) | |||
| Yield (%) | dr | Yield (%) | dr | |||||
| 1 | None | 100 | 70 (74)b,c | 95:5 | 30 | 74:26 | 26 | |
| 2 | 5 mol% of 3a instead of 1.0 equiv. of 3a | 21 | 12 | – | 5 | – | 2 | |
| 3 | 2 mol% of fac-Ir(ppy)3 instead of 3a | 7 | 0 | – | 0 | – | 0 | |
| 4 | 1.0 equiv. of benzophenone (400 nm LEDs) instead of 3a | 0 | 0 | – | 0 | – | 0 | |
| 5 | 1.0 equiv. of 3b (420 nm LEDs) instead of 3a | 17 | 5 | – | 4 | – | 3 | |
| 6 | Toluene instead of DCE | 100 | 57 | 95:5 | 46 | 84:16 | 40 | |
| 7 | EtOAc instead of DCE | 100 | 65 | 89:11 | 35 | 76:24 | 66 | |
| 8 | MeCN instead of DCE | 10 | 7 | 86:14 | 2 | – | 5 | |
| 9 | 420 nm instead of 450 nm LEDs | 37 | 14d | – | 8 | – | 2 | |
| 10 | 1.5 equiv. of 2a | 43 | 24 | 95:5 | 19 | 72:28 | 3 | |
| 11 | No 3a | 0 | 0 | – | 0 | – | 0 | |
| 12 | No light | 0 | 0 | – | 0 | – | 0 | |
| a Unless otherwise noted, the reaction conditions were as follows: 1a (0.10 mmol), 2a (0.20 mmol), 3a (0.10 mmol), DCE (0.40 mol/L), irradiation by 450 nm LEDs under Ar at 26 ℃ for 36 h; the conversion of 1a, the yield of 4a, 5a and 6a, the dr of 4a and 5a were determined by 1H NMR spectroscopy with BrCH2CH2Br as an internal standard. DCE: 1,2-dichloroethane. b Isolated yield. c 24% conversion of 3a was observed by 19F NMR with PhCF3 as an internal standard. d 100% conversion of 3a was observed by 19F NMR with PhCF3 as an internal standard. |
||||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

