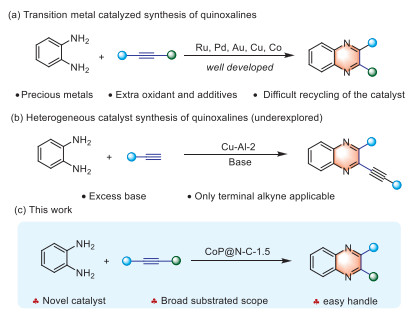
Scheme 1.
Strategies for synthesis of quinoxalines by the annulation of alkynes and 1,2-diaminobenzenes.
A novel N-stable Co2P nano-catalyst for the synthesis of quinoxalines by annulation of alkynes and 1,2-diaminobenzenes
Xiaochun Liu , Gaoyan Chen , Xiaodong Yue , Chaoyue Wang , Xue-Xin Zhang , Xuecheng Ran , Yingxiao Zong , Junke Wang , Xicun Wang

Quinoxaline and its derivatives, characterized by a double-ring system composed of benzene and pyrazine rings, represent a significant class of N-heterocyclic compounds. These compounds are extensively utilized in various fields, including natural products [1,2], pharmaceuticals [3–6], organic semiconductors [7,8], and photoelectric materials [9,10]. Consequently, the synthesis of quinoxaline structures is of considerable importance.
Currently, numerous synthesis methods have been developed [11–15]. The annulation of alkynes with 1,2-diaminobenzenes to produce quinoxalines is considered the most economical due to its cost-effectiveness and the readily available raw materials. Among these, homogeneous catalysts based on Ru, Pd, Au, Cu, and Co have garnered significant attention for their role in the synthesis of quinoxaline (Scheme 1a) [16–20]. Iodine or bromine-mediated methods for the preparation of quinoxalines have also been reported [21–23]. However, these methods often require harsh conditions, including the use of precious metal catalysts, equivalent additives, extra oxidants, and the catalyst cannot easy to recycle. These conditions result in poor functional group tolerance, low atomic efficiency and catalytic efficiency, and high production costs, thereby limiting their industrial scalability. In response to growing demands for green chemistry and sustainable development, there has been increasing interest in environmentally friendly and cost-effective non-precious metal catalysts [24,25].
Additionally, heterogeneous catalysts have garnered attention due to their ease of separation, low production costs, and environmental benefits [26–28]. The framework of heterocyclic compounds is commonly employed to construct drug molecules, natural products, fine chemicals, and functional materials [29–33]. Consequently, the application of heterogeneous catalysts in the synthesis and modification of heterocyclic compounds has garnered significant attention [34–37].
Currently, heterogeneous catalysts primarily synthesize quinoxalines using metal oxides. For instance, a copper-alumina (Cu-Al-2) catalyst was prepared via coprecipitation and calcination for the oxidative coupling of o-phenylenediamine with terminal alkynes to produce quinoxaline derivatives (Scheme 1b) [38]. Such reactions can also be catalyzed by a coordinated layer-by-layer assembly between nanosheets of reduced graphene oxide and Fe3O4@Cu2O (Fe3O4@Cu2O-rGO) [39]. The downside of these methods is that it requires excess base and only terminal alkyne applicable. Nano-catalysts, which surpass conventional catalysts in many aspects, are considered to be the most promising catalysts for the future. Their high specific surface area and abundance of active sites make them particularly valuable in various fields, including biofuels [40], organic synthesis [41], pharmaceuticals [42], and fine chemicals [43]. Nonetheless, there are few reports on the application of nano-catalysts in quinoxaline synthesis.
Our group strives to develop sustainable catalytic systems, and made notable advancements in the field of self-assembled catalysis [44] and single-atom catalyst [45]. Herein, we report the synthesis of a novel N-stabilized Co2P nano-catalyst (Scheme 1c). This catalyst exhibits a broad substrate scope and good functional group tolerance in quinoxaline synthesis, achieving yields of up to 84%. Furthermore, this catalyst is recyclable up to three times. Analysis of characterization data and experimental results revealed that the active site of the catalyst is the N-stabilized Co2P nanoparticles.
CoP@N—C-X was synthesized through a template-sacrificial approach, as illustrated in Fig. 1. Initially, melamine, cobalt phytate (PA-Co), and magnesium oxide were thoroughly mixed in ethanol. After solvent removal, the resulting Co-PA-N-MgO was subjected to pyrolysis at 800 ℃ under a nitrogen (N2) atmosphere, yielding CoP@N—C-MgO-X (where X denotes the amount of cobalt phytate). Subsequently, N-stabilized Co2P nano-catalysts were produced via pickling, resulting in three distinct catalysts: CoP@N—C-1.0, CoP@N—C-1.5, and CoP@N—C-2.0. For comparison, Co3O4@P-C was synthesized without melamine.
X-ray powder diffraction (XRD) analysis was conducted to investigate the structural properties of the catalysts (Fig. 2a). The results confirmed that the characteristic Co2P (JCPDS No. 32–0306) patterns are present in the CoP@N—C-1.0, CoP@N—C-1.5, and CoP@N—C-2.0 catalysts, with CoP@N—C-1.0 also exhibiting the characteristic peak of Co3O4 (JCPDS No. 1–078–1969) [46–48]. These findings suggest that the likelihood of forming Co2P nanoparticles increases with the rising cobalt phytate content. Additionally, the Co3O4@P-C catalyst only displays the Co3O4 characteristic peak, indicating that the introduction of nitrogen element promotes the formation of Co2P nanoparticles and inhibits the formation of Co3O4. The presence of Co and P was further validated by inductively coupled plasma optical emission spectrometry (ICP-OES), revealing 35 wt% Co and 9.6 wt% P in CoP@N—C-1.5. Comparing the Co and P contents in Co3O4@P-C with those in CoP@N—C-X indirectly suggests that nitrogen incorporation enhances the stability of Co and P elements (For details data, see Table S1 in Supporting information).
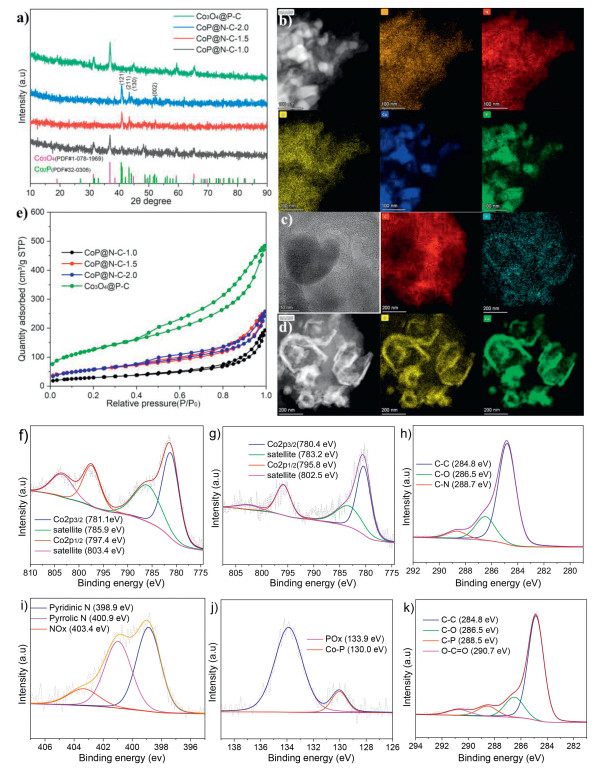
The morphologies and element distributions of the CoP@N—C-1.5 and Co3O4@P-C catalysts were characterized. The SEM images reveal that both catalysts exhibit irregular pores (For details, see Fig. S1 in Supporting information). TEM images (Fig. 2c and Fig. S2a in Supporting information) further confirm the presence of nanoparticles. Element mapping through energy-dispersive X-ray spectroscopy (EDS) demonstrates that N and O elements in the CoP@N—C-1.5 catalyst are uniformly dispersed on the carbon support, while Co and P elements aggregate to form nanoparticles (Fig. 2b and Fig. S2b in Supporting information). In contrast, in the Co3O4@P-C catalyst, the P element is uniformly dispersed on the carbon support, with Co and O forming nanoparticles (Fig. 2d). These observations are consistent with the XRD results.
Raman spectroscopy was conducted to analyze the carbon structure of the catalysts (Fig. S3a in Supporting information). The results indicated that the graphitization degree of CoP@N—C-1.5 was higher than that of CoP@N—C-1.0, CoP@N—C-2.0, and Co3O4@P-C. Additionally, N2 physisorption experiments were carried out to evaluate the pore structure and surface area of the catalysts (Fig. 2e and Fig. S3b in Supporting information). The nitrogen adsorption isotherms of CoP@N—C-X and Co3O4@P-C exhibited type Ⅳ curves with H3 hysteresis loops, confirming that the catalysts are mesoporous materials. According to the Brunauer-Emmett-Teller (BET) method, the specific surface area of CoP@N—C-1.5 was determined to be 199.93 m2/g. The Barrett-Joyner-Halenda (BJH) method was used to calculate the average pore size and pore volume, which were found to be 9.52 nm and 0.399 cm3/g, respectively (For details, see Table S2 in Supporting information).
The valence state of cobalt and the chemical environment within the catalyst were examined using X-ray photoelectron spectroscopy (XPS), with cobalt phytate serving as a reference (Fig. S4 in Supporting information). The Co 2p3/2 peak in cobalt phytate was observed at 782.1 eV In contrast, the Co 2p3/2 peak in CoP@N—C-X and Co3O4@P-C was located at 780.4–781.4 eV, indicating that the valence state of Co in these catalysts is between +2 and +3 (Figs. 2f and g, Fig. S5 in Supporting information) [49]. The C 1s spectrum of CoP@N—C-X (including CoP@N—C-1.0, CoP@N—C-1.5, and CoP@N—C-2.0) could be deconvoluted into three peaks: C—C (284.8 eV), C—O (286.4–286.5 eV), and C—N (288.5–288.7 eV) (Fig. 2h and Fig. S6 in Supporting information) [50,51]. The N 1s spectrum could be deconvoluted into three peaks as well: Pyridinic N (398.6–398.9 eV), Pyrrolic N (400.6–400.9 eV), and NOx (402.8–403.5 eV) (Fig. 2i and Fig. S7 in Supporting information) [52–55]. The P 2p spectrum revealed two peaks corresponding to POx (133.5–133.9 eV) and Co-P (129.8–130.0 eV) (Fig. 2j and Fig. S8 in Supporting information). For Co3O4@P-C, the C 1s spectrum could be deconvoluted into four peaks: C—C (284.8 eV), C—O (286.5 eV), C-P (288.5 eV), and O—C=O (290.7 eV) (Fig. 2k) [47,48]. However, due to the low phosphorus content in Co3O4@P-C, the P 2p peaks were too weak to allow for a precise determination of its chemical environment.
After elucidating the structure and active sites of the catalyst, we applied this catalyst to the annulation reaction of phenylacetylene and o-phenylenediamine was selected as a model reaction to further evaluate the relationship between the structure and performance of the synthesized catalysts (Table 1). Initially, the synthesized CoP@N—C-X catalysts (CoP@N—C-1.0, CoP@N—C-1.5, and CoP@N—C-2.0) all exhibited catalytic activity towards this reaction, whereas Co3O4@P-C showed negligible activity (entries 1–4). X-ray diffraction (XRD) analysis revealed that CoP@N—C-1.0, CoP@N—C-1.5, and CoP@N—C-2.0 contain Co2P nanoparticles, while Co3O4@P-C contains only Co3O4 nanoparticles, indicating that the Co2P nanoparticles serve as the catalytic center. Furthermore, when cobalt phytate or melamine was used as the catalyst, they demonstrated negligible activity in the reaction (entries 5 and 6). However, the use of PA-Co/N as a catalyst resulted in a small amount of the product (entry 7), suggesting that catalytic activity is only generated when N, P, and Co are simultaneously present in the catalyst. Based on these findings, The active Co2P nanoparticles in the catalyst may be stabilized by the nitrogen element.
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Catalyst | Yield (%)b |
| 1 | CoP@N—C-1.0 | 18 |
| 2 | CoP@N—C-1.5 | 58 |
| 3 | CoP@N—C-2.0 | 53 |
| 4 | Co3O4@P-C | – |
| 5 | Cobaltous phytate | Trace |
| 6 | Melamine | – |
| 7 | PA-Co/N | 12 |
| 8c | CoP@N—C-1.5 | 82 |
| a Conditions: 1a (0.2 mmol), 2a (3 equiv.) and catalyst (20 mg), DMSO (5 mL), 80 ℃ for 9 h. b Isolated yield. c At 120 ℃ for 5 h. |
||
Given the CoP@N—C-1.5 was the most effective catalyst, we further optimized the reaction conditions (Table S3 in Supporting information). Various solvents, including DMSO, toluene, methanol, 1,2-dichloroethane, and THF were tested (entries 1–5). The results indicated that the choice of solvent significantly affected the reaction outcome. DMSO emerged as the most effective solvent, achieving a yield of 58%. Following this, the reaction temperature, reactant ratio, and reaction time were systematically optimized (entries 6–12). The optimal conditions were successfully determined.
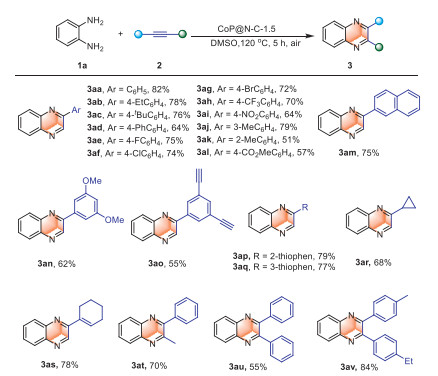
Having established the optimized condition, the generality of annulation of alkynes and 1,2-diaminobenzene with differently substituted aryl-substituted terminal was investigated (Scheme 2). The introduction of electron-donating groups allowed the reaction to proceed smoothly, producing the corresponding quinoxaline derivatives with 64%−82% yields (3aa-3ad). The introduction of electron-withdrawing groups to aryl-substituted alkynes led to the formation of quinoxaline derivatives, which yielded of 64%−75% (3ae-3ai). Notably, substrates with electron-withdrawing groups exhibited slightly lower yields, which may be attributed to a reduction in electron density of the carbon-carbon triple bonds, thereby hindering alkyne activation [56]. The yield of quinoxaline derivatives was slightly higher for 3-ethynyl-toluene (3aj) compared to 2-ethynyl-toluene (3ak), likely due to the steric hindrance. In the presence of an ester group and naphthalene, the corresponding quinoxaline can be obtained (3al, 3am). When two substituents were attached to the aryl-substituted alkyne, the corresponding products were also achieved (3an, 3ao). Additionally, when the terminal alkynes substituted by thiophene, cycloalkyl and cyclohexene, products of moderate yield can be obtained (3ap-3as). When two substituents were attached to the alkyne, the yields were 55%−84% (3at-3av). Realizing the cyclization reaction of endyne using traditional catalysts is challenging.
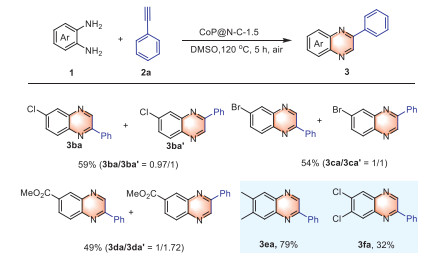
Subsequently, the range of substrates for o-phenylenediamine 1 was examined (Scheme 3). Substituted o-phenylenediamine at the 3-position produced two products due to regioselectivity (3ba-3da, 3ba'−3da'). The use of 3,5-substituted o-phenylenediamine yielded the corresponding quinoxaline derivatives (3ea, 3fa).
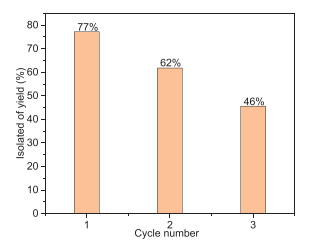
The stability of CoP@N—C-1.5 was evaluated through recycling experiments. After three cycles, the yield of quinoxaline decreased to 46% (Fig. 3), and traces of o-phenylenediamine were still detectable. Moreover, the stability and the heterogeneity of the CoP@N—C-1.5 could be demonstrated by the hot filtration experiment, in which the reaction could not proceed after the separation of the catalyst (Fig. S10 in Supporting information). X-ray diffraction (XRD) analysis of the used CoP@N—C-1.5 revealed that the crystal structure of the active site remained largely unchanged (Fig. S11 in Supporting information). The Co content and P decreased slightly (31.8% and 7.1%), as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). Transmission electron microscopy (TEM) images and energy-dispersive X-ray spectroscopy (EDS) of the used CoP@N—C-1.5 were consistent with those of the fresh CoP@N—C-1.5 (Figs. S12a-c in Supporting information). X-ray photoelectron spectroscopy (XPS) analysis showed that the chemical environment of the C and P elements remained stable (Figs. S13a and b in Supporting information). The peak corresponding to -NOx in the N 1s spectrum disappeared (Fig. S13c in Supporting information), and a new peak appeared at 778.2 eV in the Co 2p spectrum (Fig. S13d in Supporting information), attributed to Co0 [57]. This shift suggests that the observed decrease in catalyst activity may be due to changes in the chemical environment of N and alterations in the valence state of Co.
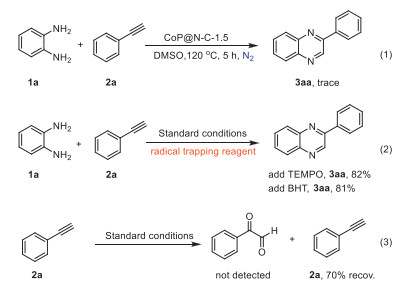
To investigate the reaction mechanism, controlled experiments were performed (Scheme 4). the reaction was nearly inactive under a nitrogen atmosphere, illustrating the crucial role of oxygen. To determine whether the reaction proceeds through a radical or ionic mechanism, radical scavenger experiments were conducted. The addition of 2 equiv. of 2,2,6,6-tetramethylpiperidin-1-yl-oxyl (TEMPO) or 3,5-di‑tert-butylhydroxytoluene (BHT) to the reaction system had small impact on the product yield. This suggest that the reaction is less likely to undergo radical pathway. To determine if the reaction proceeds via the glyoxal mechanism, we performed experiment [20]. No glyoxal compounds were detected by thin-layer chromatography (TLC) and gas chromatography (GC). Additionally, the recovery yield of 70% for phenylacetylene, attributable to its volatility, suggests that the reaction does not follow the glyoxal mechanism.
In summary, we have designed and synthesized a novel nano-catalyst (CoP@N—C-1.5), and used it to catalyze the annulation reaction of alkynes and 1,2-diaminobenzenes. The reaction showed a broad substrate scope and good functional group tolerance allowing the synthesis of a wide range of quinoxalines in good to high yields and the catalyst can be recycled up to three times. The active site of this catalyst consists of Co2P nanoparticles stabilized by nitrogen, as confirmed through structural characterization and experimental analysis. Detailed investigation into the reaction mechanism is currently underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xiaochun Liu: Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Gaoyan Chen: Validation, Investigation, Formal analysis, Data curation. Xiaodong Yue: Validation, Investigation. Chaoyue Wang: Validation, Investigation. Xue-Xin Zhang: Writing – review & editing, Visualization. Xuecheng Ran: Validation. Yingxiao Zong: Supervision, Resources, Funding acquisition. Junke Wang: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition. Xicun Wang: Writing – review & editing, Supervision, Investigation.
The authors gratefully acknowledge support from the National Natural Science Foundation of China (Nos. 22061017 and 21862006), and the Science and Technology program of Gansu Province (Nos. 22YF7GG127 and 23JRRG0002), National innovative training program for college students (No. 202310740015), the Open Project Funding of Hubei Key Laboratory of Processing and Application of Catalytic materials (No. 202306004).
Supplementary material associated with this article can be found, in the online version, at doi:
A.K. Parhi, Y. Zhang, K.W. Saionz, et al., Bioorg. Med. Chem. Lett. 23 (2013) 4968–4974.
A. Zeb, A. Hameed, L. Khan, et al., Med. Chem. 10 (2014) 724–729. doi: 10.2174/1573406410666140526145429
S.T. Hazeldine, L. Polin, J. Kushner, et al., J. Med. Chem. 44 (2001) 1758–1776.
R.A. Smits, H.D. Lim, A. Hanzer, et al., J. Med. Chem. 51 (2008) 2457–2467. doi: 10.1021/jm7014217
Y.B. Kim, Y.H. Kim, J.Y. Park, S.K. Kim, Bioorg. Med. Chem. Lett. 14 (2004) 541–544.
S. Tariq, K. Somakala, M. Amir, Eur. J. Med. Chem. 143 (2018) 542–557.
S. Ammermann, C. Hrib, P.G. Jones, et al., Org. Lett. 14 (2012) 5090–5093. doi: 10.1021/ol302348v
L. Wang, W. Guo, X.X. Zhang, X.D. Xia, W.J. Xiao, Org. Lett. 14 (2012) 740–743. doi: 10.1021/ol203275b
W. Wang, Y. Shen, X. Meng, et al., Org. Lett. 13 (2011) 4514–4517. doi: 10.1021/ol201664x
J. Yuan, J. Ouyang, V. Cimrová, et al., J. Mater. Chem. C 5 (2017) 1858–1879.
H. Khatoon, E. Abdulmalek, Molecules 26 (2021) 1055. doi: 10.3390/molecules26041055
V.K. Maikhuri, A.K. Prasad, A. Jha, S. Srivastav, New J. Chem. 45 (2021) 13214–13246. doi: 10.1039/d1nj01442k
L. Biesena, T.J.J. Müller, Adv. Synth. Catal. 363 (2021) 980–1006.
J. Gao, Z.G. Ren, J.P. Lang, Chin. Chem. Lett. 28 (2017) 1087–1092.
A. Kumar, T.M. Dhameliya, R.V. Hirani, A.J. Bhatt, J. Mol. Struct. 1259 (2022) 132732.
Y. Xu, X. Wan, Tetrahedron Lett. 54 (2013) 642–645.
S. Shi, T. Wang, W. Yang, M. Rudolph, A.S.K. Hashmi, Chem. Eur. J. 19 (2013) 6576–6580. doi: 10.1002/chem.201300518
S.A. Jadhav, A.P. Sarkate, M.G. Shioorkar, D.B. Shinde, Synth. Commun. 47 (2017) 1661–1667. doi: 10.1080/00397911.2017.1337153
C.F. Gers-Panther, H. Fischer, J. Nordmann, et al., J. Org. Chem. 82 (2016) 567–578.
H.R. Yang, Z.Y. Hu, X.C. Li, L. Wu, X.X. Guo, Org. Lett. 24 (2022) 8392–8396. doi: 10.1021/acs.orglett.2c03465
J. Zi, D.W. Gu, Y. Zhang, et al., Synth. Commun. 48 (2018) 915–920. doi: 10.1080/00397911.2018.1428752
W. Zhang, X. Yang, Z. Cao, et al., ChemistrySelect 8 (2023) e202302634.
A. Das, K.R.J. Thomas, Asian J. Org. Chem. 9 (2020) 1820–1825. doi: 10.1002/ajoc.202000388
J.V. Obligacion, P.J. Chirik, Nature Rev. Chem. 2 (2018) 15–34. doi: 10.1038/s41570-018-0001-2
P. Gandeepan, T. Müller, D. Zell, et al., Chem. Rev. 119 (2019) 2192–2452. doi: 10.1021/acs.chemrev.8b00507
W. Fang, T. Tu, Sci. China Chem. 66 (2023) 299–300. doi: 10.1007/s11426-022-1457-1
W.J. Li, Z.Q. Wang, J.B. Wang, et al., J. Catal. 430 (2024) 115308.
F. Ghaffarian, M.A. Ghasemzadeh, S.S. Aghaei, J. Mol. Struct. 1186 (2019) 204–211.
Q.H. Liu, S.L. Kang, Z.S. Cui, et al., Green Chem. 26 (2024) 4803–4810. doi: 10.1039/d4gc00502c
J. Safaei-Ghomi, M.A. Ghasemzadeh, A. Kakavand-Qalenoei, J. Saudi Chem. Soc. 20 (2016) 502–509. doi: 10.1016/j.jscs.2012.07.010
R. Bakhshali-Dehkordi, M.A. Ghasemzadeh, J. Safaei-Ghomi, J. Mol. Struct. 1216 (2020) 127698.
J.S. Ghomi, S. Zahedi, M.A. Ghasemzadeh, Monatsh. Chem. 145 (2014) 1191–1199.
J.S. Ghomi, S. Zahedi, M.A. Ghasemzadeh, Res. Chem. Intermed. 41 (2015) 8625–8636.
H.Y. Song, J. Jiang, C. Wu, et al., Green Chem. 25 (2023) 3292–3296. doi: 10.1039/d2gc04843d
Y.H. Lu, C. Wu, J.C. Hou, et al., ACS Catal. 13 (2023) 13071–13076. doi: 10.1021/acscatal.3c02268
S. Mekki, Y. Laichi, F. Benourrad, et al., Appl. Organomet. Chem. 37 (2023) e7103.
A. Bharathi, S.M. Roopan, A. Kajbafvala, et al., Chin. Chem. Lett. 25 (2014) 324–326.
A.V. Nakhate, K.B. Rasal, G.P. Deshmukh, S.S.R. Gupta, L.K. Mannepalli, J. Chem. Sci. 129 (2017) 1761–1769. doi: 10.1007/s12039-017-1393-0
Z. Wang, G. Hu, J. Liu, et al., Chem. Comm. 51 (2015) 5069–5072.
H. Ye, J. Shi, Y. Wu, et al., Fuel 356 (2024) 129594.
M. Ashraf, M.S. Ahmad, Y. Inomata, et al., Coord. Chem. Rev. 476 (2023) 214928.
J. Hou, M. Kazemi, Res. Chem. Intermed. 50 (2024) 1713–1743. doi: 10.1007/s11164-024-05243-3
B. Maleki, H. Esmaeili, Renew Energy 209 (2023) 10–26.
J. Wang, Y. Zong, X. Wang, et al., Green Chem. 18 (2016) 967–973.
X. Liu, C. Wang, J. Meng, et al., Chin. Chem. Lett. 34 (2023) 108745.
B. You, N. Jiang, M. Sheng, et al., Chem. Mater. 27 (2015) 7636–7642. doi: 10.1021/acs.chemmater.5b02877
H. Liu, J. Guan, S. Yang, et al., Adv. Mater. 32 (2020) 2003649.
J. Song, C. Zhu, B.Z. Xu, et al., Adv. Energy Mater. 7 (2017) 1601555.
L. Huang, H. Zhang, Y. Cheng, et al., Chin. Chem. Lett. 33 (2022) 2569–2572.
J.M. Stillahn, K.J. Trevino, E.R. Fisher, ACS Appl. Mater. Interfaces 3 (2011) 1402–1410. doi: 10.1021/am101282y
H.H. Wang, N. Leaukosol, Z.B. He, P. Navard, Cellulose 20 (2013) 1587–1601. doi: 10.1007/s10570-013-9945-z
X. Li, Y. He, S. Cheng, et al., Adv. Mater. 33 (2021) 2106371.
D. Xi, J. Li, J. Low, et al., Adv. Mater. 34 (2022) 2104090.
M. Li, H. Wang, W. Luo, et al., Adv. Mater. 32 (2020) 2001848.
X. Hu, D. Zhou, H. Wang, et al., Chin. Chem. Lett. 34 (2023) 108050.
H. Liu, P. Zhu, D. Yang, et al., J. Am. Chem. Soc. 146 (2024) 2132–2140. doi: 10.1021/jacs.3c11632
Q. Zhu, F. Wang, F. Zhang, Z. Dong, Nanoscale 11 (2019) 17736–17745. doi: 10.1039/c9nr04867g

Scheme 1 Strategies for synthesis of quinoxalines by the annulation of alkynes and 1,2-diaminobenzenes.

Figure 2 (a) XRD of CoP@N—C-1.0, CoP@N—C-1.5, CoP@N—C-2.0 and Co3O4@P-C. (b) Elemental mapping image of CoP@N—C-1.5. (c) TEM image of Co3O4@P-C. (d) Elemental mapping image of Co3O4@P-C. (e) Nitrogen adsorption isotherm of CoP@N—C-1.0, CoP@N—C-1.5, CoP@N—C-2.0 and Co3O4@P-C. (f) Co 2p XPS spectrum of CoP@N—C-1.5. (g) Co 2p XPS spectrum of Co3O4@P-C. (h) C 1s XPS spectrum of CoP@N—C-1.5. (i) N 1s XPS spectrum of CoP@N—C-1.5. (j) P 2p XPS spectrum of CoP@N—C-1.5. (k) C 1s XPS spectrum of Co3O4@P-C.

Scheme 2 Substrate scope. Conditions: 1a (0.2 mmol), 2 (3 equiv.), CoP@N—C-1.5 (20 mg) at 120 ℃ in DMSO (5 mL) for 5 h, unless otherwise noted. Yield of isolated products.

Scheme 3 Substrate scope. Conditions: 1 (0.2 mmol), 2a (3 equiv.), CoP@N—C-1.5 (20 mg) at 120 ℃ in DMSO (5 mL) for 5 h in the air. Yield of isolated products.
Table 1. Optimization of catalysts.a
|
||
| Entry | Catalyst | Yield (%)b |
| 1 | CoP@N—C-1.0 | 18 |
| 2 | CoP@N—C-1.5 | 58 |
| 3 | CoP@N—C-2.0 | 53 |
| 4 | Co3O4@P-C | – |
| 5 | Cobaltous phytate | Trace |
| 6 | Melamine | – |
| 7 | PA-Co/N | 12 |
| 8c | CoP@N—C-1.5 | 82 |
| a Conditions: 1a (0.2 mmol), 2a (3 equiv.) and catalyst (20 mg), DMSO (5 mL), 80 ℃ for 9 h. b Isolated yield. c At 120 ℃ for 5 h. |
||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


