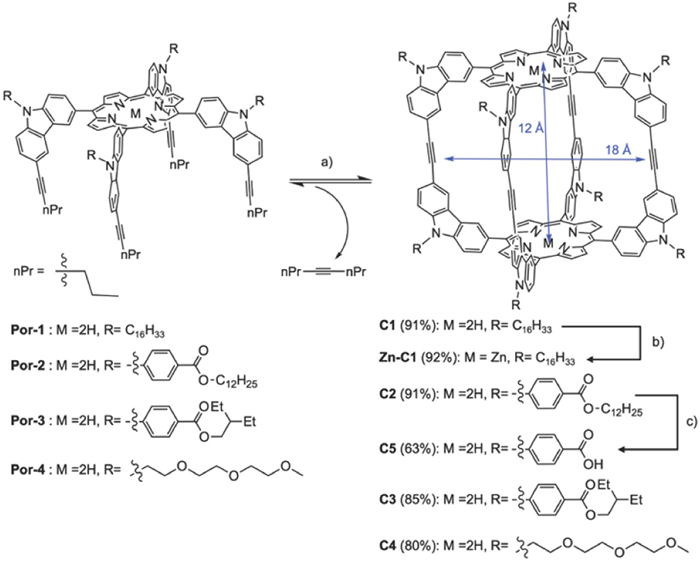
Scheme 1.
Synthesis of molecular cages. Conditions: (a) Mo catalyst, CHCl3, 5 Å molecular sieves, reflux; (b) Zn(OAc)2, CHCl3/MeOH, reflux; (c) KOH, THF/MeOH/H2O, reflux.
Artificial transmembrane channel constructed from shape-persistent covalent organic molecular cages capable of ion and small molecule transport
Brandon Bishop , Shaofeng Huang , Hongxuan Chen , Haijia Yu , Hai Long , Jingshi Shen , Wei Zhang

Natural transmembrane channels play a vital role in a wide range of biological functions, including cellular communication [1], signal transduction [2-4], and the maintenance of osmotic pressure and pH [5-7]. Defects in natural transmembrane channels can result in a class of medical conditions termed channelopathies, examples of which include cystic fibrosis and epilepsy [8,9]. Artificial systems for transmembrane transport could potentially serve as treatments for such disorders through channel replacement therapies. Although several synthetic transmembrane channels or carriers have been discovered that can mimic their biological counterparts, maintaining their functionality in a biological environment remains a challenge, limiting their industrial and biomedical applications [10-13]. In recent years, numerous artificial transmembrane transporters have been developed from small organic molecules [14-17], macrocyclic compounds [18-27], metal-organic frameworks (MOFs) [28-31], polymers [32-38], peptide mimics [39-43], foldamers [44-47], and molecular machines, such as molecular shuttles or molecular swings [48-50]. These structures primarily rely on supramolecular interactions such as metal-coordination bonding, π–π stacking, hydrophobic interactions, electrostatic forces, and hydrogen bonding to form channel-like structures [51,52]. These relatively weak non-covalent interactions can pose challenges in precisely controlling the self-assembly of individual molecules and can compromise their stability in biological environments [53,54]. Owing to the limitations of supramolecular structures, the development of discrete monomeric transmembrane channels constructed from covalent molecules has garnered interest. Such ion channels offer precise control over structure and function, which would be challenging to achieve using a supramolecular approach.
Since the first example in 2009, well-defined, purely organic cages connected by covalent bonds, known for their high chemical and thermal stability and referred to as covalent organic polyhedrons (COPs) [55-57], have garnered significant attention because of their unique properties and broad applications. Various COPs have been developed for applications such as host-guest chemistry [58,59], gas adsorption [60-62], chemical sensing [63,64], catalysis [65-67], and nanoparticle growth [68,69]. Kim and coworkers pioneered the use of a series of imine-linked COPs as monomeric transmembrane transporters for selectively transporting iodide and glucose [70,71]. This approach could potentially be used to sense cells with knocked-out genes. However, direct cell-imaging leveraging the internal cavity of the COPs remains an uncharted territory, and the structure-property relationship guiding the COPs' design is still not fully understood. It's worth noting that most biological systems operate in an aqueous environment, so the chemical resistance of these structures towards hydrolysis should also be considered. Over the past decade, our group has successfully synthesized multiple shape-persistent arylene-ethynylene COPs via dynamic alkyne metathesis [72-74], achieving high yields. The resulting COPs, which feature good solubility, functional group tunability, rigid backbones, accessible internal cavities, and robust linkages, are potentially well-suited as candidates for transmembrane channels.
Herein, we designed and synthesized a series of porphyrin-based, shape-persistent arylene-ethynylene COPs via one-step alkyne metathesis. These COPs have the same backbone but feature functional groups of varying lengths and polarities on eight vertices. These groups form unimolecular channels in lipid bilayer membranes, transporting protons with varying efficiencies. We observed reduced transportation efficiency when we intentionally altered the electronic environment of the channel via metalation of the porphyrin panels (with Zn2+). Furthermore, when we deliberately blocked the internal cavity of the cage (with C70), the transmembrane transport activity was completely terminated. Both of these observations highlight the importance of the void space inside the cage-based transporters. Molecular dynamics simulations were used to model the conformation of the COPs when inserted into lipid bilayers, confirming the monomeric nature of these transporters. Additionally, transportation of calcein was made possible due to the large cavity of the COPs, and direct cell-imaging in a living-cell system was demonstrated.
A series of porphyrin-based rectangular prism shaped-persistent cages with a large internal void (1.8 × 1.2 nm) were synthesized. Substituents with different lengths and polarities were installed on eight vertices. A highly active multidentate Mo(Ⅵ) complex was used as the metathesis catalyst [75]. Utilizing adsorption-driven alkyne metathesis for cage formation resulted in high isolation yields of 80%−91% (Scheme 1). The zinc-metalated cage (Zn-C1) was prepared by reacting C1 with Zn(OAc)2. The carboxylic acid functionalized cage C5 was obtained from the hydrolysis of its ester derivative C3.
We used a pH-sensitive dye, 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) as the fluorescence probe to evaluate the channeling activities of the cages. HPTS-encapsulated large unilamellar vesicles (LUVs) or liposomes were prepared as previously reported from L-α-phosphatidylcholine and HPTS [76]. The aqueous interior of the LUVs was buffered at pH 7.0 (10 mmol/L HEPES, 100 mmol/L NaCl, ~0.1 mmol/L HPTS), and the exterior was initially buffered at pH 7.0 (10 mmol/L HEPES, 100 mmol/L MX, M = Na+, Li+, K+, Rb+, or Cs+, X = Cl− or Br−). A solution of one of the cages in organic solvent (1.0 mmol/L; C1, Zn-C1, C2, C3, C5: THF, C4: DMF) was then added into the above suspension of HPTS-encapsulated LUVs and incubated for 3 min. After the incubation period, 1.0 mol/L NaOH was added (4 µL) to generate the pH gradient between the intravesicular and extravesicular buffers. The fluorescence emission of the dye was immediately monitored at room temperature for 500 s.
After 480 s, Triton X-100, a surfactant, was added to completely disrupt the LUVs and achieve the maximal fluorescence emission of HPTS as the 100% reference point. As shown in Fig. 1B, when the exterior buffer solution consists of NaCl (M = Na+, X = Cl−), rapid increase of HPTS fluorescence emission was observed in the presence of 20 µmol/L (1.5 mol% relative to lipid) of C1, which reaches 70% of the maximal level within 500 s. Such increase of HPTS fluorescence suggests possible proton efflux or hydroxide influx in response to the transmembrane pH gradient (∆pH = 0.6), thus the increase of intravesicular pH. In order to confirm the fluorescence intensity increase is associated with the presence of the cage, we performed a blank experiment in the absence of the cage under otherwise identical conditions. A very small increase (≤10%) of fluorescence intensity was observed in the blank experiment even after 480 s. This supports that the cage is indeed responsible for the observed increase in fluorescence intensity. As low as 2.5 µmol/L (0.38 mol% relative to lipid) of C1 could reach 45% maximal fluorescence intensity within 500 s (Fig. 1B). Such activity of C1 is comparable to many reported synthetic ion channels. A Hill Analysis was performed in order to gain more insight into the transport behavior. The analysis confirmed that the cage can form a highly active transmembrane channel with an EC50 (the effective monomer concentration needed to reach 50% activity) equal to 5.17 µmol/L (0.75 mol% relative to lipid) [77,78]. In order to confirm the integrity of the LUVs was maintained upon exposure to the cages, dynamic light scattering (DLS) measurements were performed on the vesicles before and after being exposed to the cage. The data confirmed that the membranes were not disturbed and that the size of the vesicles only increased slightly (< 5%), which may indicate successful insertion of the cage into the lipid bilayer (Fig. S1 in Supporting information).

We observed significantly slower increase of HPTS fluorescence emission when the extravesicular cation was changed from Na+ to K+ (Fig. 1C). This suggests that cations are involved in the ion transport process. Since our HPTS assay is sensitive to external cation change, the cage likely mediates cation antiport (i.e. H+/M+). To further investigate, we also monitored proton transport with the Li+, Rb+, and Cs+ as the external cations. As expected, when the external cation was Li+ the proton transport increased, reaching over 75% fluorescence intensity within 500 s with the same cage loading (Fig. 1C). Furthermore, as the cation size increased the maximal fluorescence intensity reached decreased as shown in Fig. 1C. When the external cation was Rb+ with the same loading of C1 much lower transport efficiency was observed, showing approximately 25% increase in fluorescence intensity. This decreased further to less than 10% increase in fluorescence when the external cation was Cs+. This further supported the idea that the mechanism of transport is cation antiport (i.e., H+/M+). Anion antiport of OH−/X− and the osmotically disfavored unidirectional symports of H+/X− or M+/OH− are less likely. In order to further confirm the cation transport selectivity of the cage, we performed the HPTS assay in the presence of a different anion, bromide. Wu and Gale demonstrated that the 'dual gradient' method is unreliable for anions when cation transport is rate limiting [79]. Therefore, the anion was changed to bromide in both the exterior and interior buffer solutions. No significant change in the rate of increase of fluorescence intensity was observed with the anion replacement of Cl− with Br− (M+ = Na+ in both cases) as shown in Fig. 1C. Such insensitivity of the HPTS assay to anion change further supports that the cage preferably transports cations (i.e., H+ efflux and M+ influx) rather than anions.
In order to further confirm the transport mechanism of the cage and examine the proton transport selectivity of the cage, we also monitored the combined action of the potassium carrier valinomycin and C1 on the decay of the transmembrane pH difference in the HPTS assay with KCl as the external salt. We found that in the absence of cage, valinomycin (0.2 mol%, relative to lipid) exhibits low activity, showing only around 20% increase of fluorescence intensity within 500 s (Fig. 2A). However, the combination of valinomycin and C1 (20 µmol/L) significantly enhances the ion transport rate, reaching over 80% of HPTS fluorescence emission within 500 s (Fig. 2A), which is approximately a two-fold increase in fluorescence intensity compared to the case of the HPTS assay in the presence of C1 alone (45%, Fig. 2A). These results indicate that the rate limiting step of the H+/K+ antiport mediated by the cage is not the proton efflux but the disfavored influx of K+, supporting the proton selectivity of such cage-based ion channel. This trend was consistent with the Zn-metalated version of C1 (Fig. S9 in Supporting information). Furthermore, this testing was carried out with the more active cage, C2. When the external salt was KCl, C2 (20 µmol/L) only displays a 50% increase in fluorescence intensity (Fig. S10A in Supporting information). However, as seen with C1, in the presence of both valinomycin and C2, the fluorescence intensity can reach 100%. This further confirms the transport mechanism of the cage and supports that cation antiport is rate limiting.
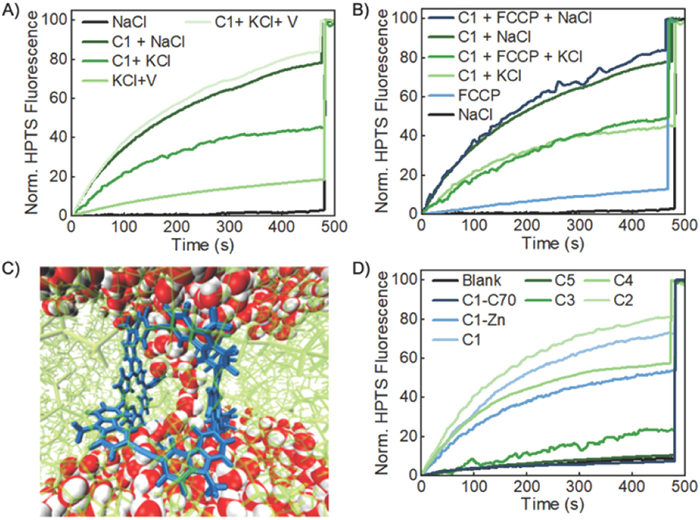
In addition to the valinomycin testing, we also confirmed that the transport of the cation was rate limiting using carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), a proton transporter, with KCl and NaCl as the external salts. We found that in the absence of the cage, FCCP (0.2 mol% relative to lipid) exhibits low activity, showing only around 10% increase of fluorescence intensity within 500 s in the presence of either external salt (Fig. 2B). Additionally, the combination of FCCP and C1 (20 µmol/L) did not significantly enhance the ion transport rate and showed only an 80 increase in fluorescence intensity when the external salt was NaCl. This is approximately a 10% increase compared to the cage alone (Fig. 2B). Likewise, when the external salt was KCl, the combination of FCCP and C1 led to a 48% increase in fluorescence intensity. This is only a 9% increase in fluorescence intensity compared to the cage alone. Similarly to the valinomycin testing, the FCCP testing was carried using C2 as a comparison. Analogously to C1, C2 did not display an increase in transport efficiency when combined with FCCP. The combination of FCCP and C2 (20 µmol/L) did not significantly enhance the ion transport rate and showed only an 82% increase in fluorescence intensity when the external salt was NaCl. This is only a 5% increase compared to the cage alone (Fig. S10B in Supporting information). Also, when the external salt was KCl, the combination of FCCP and C2 led to a 56% increase in fluorescence intensity. This is only a 7% increase in fluorescence intensity compared to the cage alone (Fig. S10B). The combination of the results from the valinomycin and FCCP testing further confirm the transport mechanism of the cage and indicate that the rate limiting step is of the H+/K+ antiport mediated by the cage is disfavored influx of K+.
|
|
(1) |
|
|
(2) |
Next, the kinetics of proton transport were investigated using C1. The kinetic analysis procedure for transmembrane channeling process reported by Regen was followed [80]. The observed rate constants kobsd were obtained from the fluorescence traces using Eq. 1, where t1/2 is the time required for the fluorescence intensity to reach 50% of the observed maximum. We assume n molecules of the cage form the active species that function as ion channels and define K as the equilibrium constant between monomer and the active channel species. Then the obtained kobsd and the corresponding cage concentration can be fitted in Eq. 2, where k represents the proton transport rate constant through the channels. As shown in Fig. 1D, the kinetic profile shows apparent first-order rate constant for the proton transport process, which can be fitted by a linear correlation (n = 1.0) with the concentration of C1. The kinetic results suggest that the active species for proton transport is likely a monomeric cage. The cages insert into the lipid bilayer membrane with the aid of long hydrophobic side chains (e.g., hexadecyl in C1), which are capable of interacting with lipid tails and long enough to span the lipid bilayer. Shorter and more polar chains that are inadequate to span the full thickness of the bilayer membrane (3–5 nm) likely lead to decreased channel efficiency due to their limited partition and transient blockage of the channel by the dynamically flowing lipids (e.g., C3 and C5) [81].
A Molecular dynamics (MD) simulation was employed to model such a structure. Fig. 2C shows the MD simulation snapshot after a 10 ns simulation. The bulk lipid bilayer has a thickness of 5 nm, consistent with the literature, but becomes as thin as 2 nm in the region with the cage [81]. The porphyrin segments of the cage are parallel to the lipid tails and stabilized by the hydrophobic interaction between them. A water chain is observed in the middle of the channel created by the cage, connecting the bulk water regions separated by the lipid bilayer. In order to examine the stability of this water chain, we further extended our simulation for another 10 ns, and found that this water channel still exists (details can be found in Supporting information). This channel-spanning water chain is consistent with what has been seen in other proton-selective transmembrane channels. Specifically, Liu and Zeng recently published a foldamer-based proton-selective channel in which they noticed a similar water chain form and span the entire channel length [44].
Interestingly, the ionophoric activities of cages are considerably influenced by the peripheral substituents. Higher activity was observed with cages containing longer and less polar alky chains (C1 and C2) (Fig. 2D). This may be due to the long alkyl chains mimicking lipid tails allowing for greater interaction with the interior of the lipid bilayer. The dodecyl-ester substituted cage, C2, had an EC50 = 2.32 µmol/L (0.35 mol% relative to lipid), which showed enhanced proton transport compared to C1. This may be due to the increased length of the sidearm, as well as an increase in ability to interact with the phospholipid. The butyl‑ester substituted cage, C3 [R = -CH2CH(Et)2)], with the same loading (20 µmol/L) shows almost no increase of HPTS fluorescence emission (< 20% after 500 s). The triethylene glycol (Tg) substituted cage, C4, shows diminished activity (EC50 = 13.02 µmol/L (1.8 mol% relative to lipid)) compared to C1 even though it possesses a long Tg chain as the sidearm. This indicates that the polarity of the sidearm is important to consider and that more polar sidearms show diminished transport activity likely due to less favorable interactions with the lipid tails. The COOH-substituted cage, C5, with even shorter and more polar side chains (R = -COOH) displays minimal channel activity, reaching only up to 15% of the maximal fluorescence regardless of the loading amount (5 µmol/L - 30 µmol/L) (Fig. 2D and Fig. S7 in Supporting information). This is in great contrast to the previously reported example of tetraporphyrin metallacycle-based ion channels, where the peripheral carboxylic acid residues were critical for the channeling activities. It has been reported that such metallacycles form dimers through hydrogen bonding and function as transmembrane nanopores when substituted with carboxylic acids, whereas their methyl ester analogs show no such activity [76]. However, on the contrary to the reported porphyrin containing metallacycles, C5 is inactive, although the formation of dimers that can span the entire depth of the membrane and function as transmembrane channels is expected. Our results therefore suggest that the active channels in the present work are not likely a dimer of the cage formed through supramolecular interactions, but rather a unimolecular channel.
It is interesting to note that the metalated analogue Zn-C1 with long alkyl substituents (R = C16H33) displays decreased proton channeling activity (EC50 = 7.87 µmol/L (0.875 mol% relative to lipid)) compared to the parent cage, C1 (Fig. 2D). When the same loading was applied (20 µmol/L), Zn-C1 reaches only 50% maximal fluorescence intensity at 500 s, whereas C1 reaches 70% maximum within 500 s. In order to understand the limiting factor of proton transport efficiency in regard to Zn-C1, we performed the HPTS assay in the presence of valinomycin (0.2 mol% relative to lipid) (Fig. S9). In the absence of Zn-C1, no significant increase of fluorescence intensity was observed in the HPTS assay in the presence of valinomycin alone. However, upon the addition of Zn-C1, rapid and dramatic increase of HPTS fluorescence emission was observed (70% of maximum), indicating the disfavored potassium influx is responsible for the observed slow proton transport by Zn-C1. Presumably, metalation of two porphyrin moieties of the cage interfere with K+ transport by electrostatic repulsion and may also have a negative effect on the insertion of the cage into hydrophobic lipid bilayer. In order to further confirm this, we also metalated C1 with nickel to obtain Ni-C1. The transport behavior of Ni-C1 was similar to the zinc metalated cage. When the same loading is applied (20 µmol/L), Ni-C1 reaches only 55% maximal fluorescence at 500 s (Fig. S4 in Supporting information).
In order to investigate whether the ions are transported through the internal cavity of the cage, we blocked the inside cavity of the cages with C70 [42]. Our previous study shows that C1 is an excellent fullerene receptor, forming a 1:1 host–guest complex with exceptionally high binding affinity (e.g., C70 Kassoc = 1.5 × 108 L/mol in toluene). Furthermore, based on previous titration experiments performed, the solubility of the cages is not affected after incorporation of C70 into the internal cavity [42]. We prepared the C70-encapsulated cage compounds as previously reported and measured its proton transport activity by the HPTS assay. As shown in Fig. 2D, C70@C1 shows almost no ionophoric activity, indicating a successful channel blockage by C70. Furthermore, none of the cages showed significant ionophoric activity when blocked by C70, supporting that the mechanism of transport is consistent between cage compounds (Fig. S8 in Supporting information). These results suggest that the internal cavity of the cages plays a critical role in the transport of ions. It also provides coherent evidence supporting our proposed monomeric channeling mechanism, in which the channeling process is mediated through the cage cavity rather than a barrel stave or carpet-like rupture.
To the best of our knowledge, this is the first report of using fullerenes to shut down synthetic transmembrane channels, thus providing another regulatory mechanism to those widely investigated ones, such as enzyme-gated [82], voltage-gated [83], ligand-gated [84], and photochemically switchable approaches [81].
Other than protons, transportation of small molecules across cell membranes has also attracted long time interest in the field of synthetic transmembrane channels/pores. Based on the computer modelling study, the dimension of the cage internal cavity is 1.8 × 1.2 nm, which is large enough to transport small molecules like calcein (a hydrated radius of approx. 0.6 nm) [85]. In order to investigate whether the cages can act as synthetic pores, we performed a calcein transport assay with calcein-encapsulated LUVs (40 mmol/L calcein, internal buffer 10 mmol/L HEPES, 100 mmol/L NaCl, pH 7.4; external buffer 10 mmol/L HEPES, 100 mmol/L NaCl, pH 7.4). As with the HPTS encapsulated LUVs, the integrity of the calcein-encapsulated LUVs was checked by dynamic light scattering (DLS) measurements. Similarly, the data confirmed that the membranes were not disturbed and that the size of the vesicles only increased slightly (< 10%), which may indicate successful insertion of the cage into the lipid bilayer (Fig. S11 in Supporting information).
After incubation for 3 min with C1, we observed steady increase of fluorescence intensity (Fig. 3A), indicating the export of calcein from the LUVs and thus reduced self-quenching. The calcein assay in the absence of cage molecules under otherwise identical conditions shows < 5% increase even after 500 s, suggesting C1 is responsible for the observed transmembrane transport of calcein. As before, a Hill Analysis was performed and revealed the EC50 for C1 to be 13.55 µmol/L (1.8 mol% relative to lipid) (Fig. S12 in Supporting information). The remaining 3 cages (Zn-C1, C2, C4) also showed varying levels of calcein transport based on their substituents. Similar to the trend seen in proton transport, the cages with longer and less polar sidearms (C1, Zn-C1, C2) displayed superior transport activity compared to the cages with more polar sidearms (C4). This further supports the proposed idea that the sidearms play a crucial role in insertion into the lipid bilayer and therefore the channeling capabilities of the cages. C2 which showed the greatest proton transport capabilities showed similar calcein transport activity (EC50 = 13.21 µmol/L (1.75 mol% relative to lipid)) when compared to C1. As before, Zn-C1 showed reduced small molecule transport (EC50 = 25.85 µmol/L (4.0 mol% relative to lipid)) compared to its free-base counterpart C1 (Fig. 3B). This may be due to unfavored interactions between the cage and the hydrophobic lipid bilayer as discussed previously.
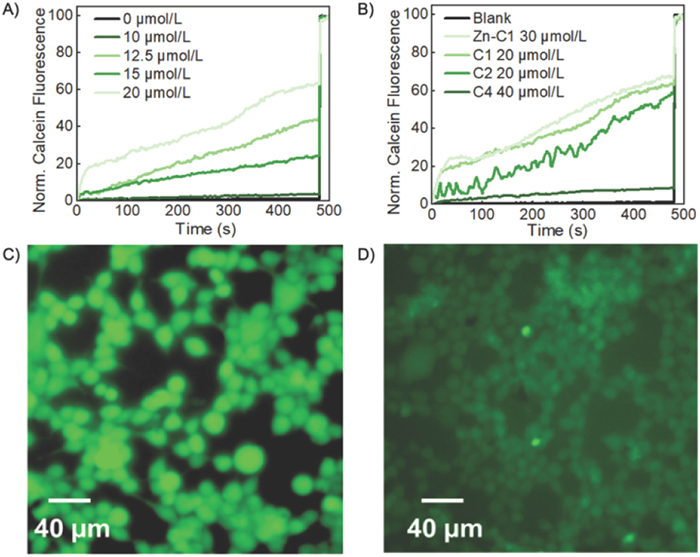
In order to further confirm the calcein transport activity of the cages, we examined the transport of calcein by C1 into human embryonic kidney (HEK) 293 T cells by fluorescence imaging. HEK 293 T cells were cultured at 37 ℃ with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The solution of C1 was added into the cell suspension. After 10 h of cell adhesion, calcein was added into the medium and incubated for a further 30 min. The free calcein was removed by repeated washing with phosphate-buffered saline (PBS) and the cells were imaged by a fluorescence microscope. As shown in Fig. 3C, for the cells with C1 supplemented, fluorescence emission was observed for majority of cells. However, in the absence of the cage (Fig. 3D), most of the cells exhibited background fluorescence. In order to confirm the dye molecules are internalized into the cells in the presence of cage molecules, we measured fluorescence of the respective cell lysates (Fig. S16 in Supporting information). Consistent with the fluorescence imaging results, the fluorescence intensity of the sample with both C1 and calcein added was significantly higher (15-fold) than that of the sample without the cage, further supporting that C1 is capable of acting as artificial channels to transport small molecules like calcein into cells. Considering that calcein can pass through the channel composed of cages, similar HPTS efflux might be possible. However, considering the large concentration of cations/anions (100 mmol/L) and their much smaller size, compared to HPTS (< 0.1 mmol/L), the transport of small ions is likely much more rapid and dominant than HPTS transport in the previously discussed HPTS assay.
Together the above evidence corroborates that the molecular cages with a rigid backbone and hydrophobic cavity are able to function as transmembrane channels via insertion into the lipid bilayer. Long alkyl side chains installed at the vertices of the cages provide hydrophobic interactions with the lipid tails of similar molecular structure and create a corridor for channeling. The cages are likely oriented with two porphyrin rings parallel to the lipids, thus exposing the cavity for the passage of ions and small molecules, as shown in Fig. 2C. Although we prefer to refer to the cages as channels, the possibility that they act as carriers cannot be ruled out. Further mechanistic studies (e.g., conductance experiment) on the ion/small molecule transport mediated by the cage will be conducted and reported in due course.
In summary, a series of porphyrin-based, shape-persistent molecular cages, substituted with various flexible side chains, have been constructed through alkyne metathesis, and their channeling activities have been investigated. We found that C1 and C2, which feature eight long alkyl side chains, function as efficient proton and small molecule channels. In contrast, whereas cages C3, C4, and C5, with shorter and/or more polar side chains, exhibit impaired channeling activity. These results suggest that hydrophobic arms, capable of interacting with lipid tails and long enough to span both ends of the lipid bilayer, are critical to the formation of an effective channel. Kinetic analysis indicates that the rate of proton transport linearly correlates with the cage concentration. This suggests that a monomeric cage likely forms the individual active channel, and the channeling mechanism is unlikely to resemble a barrel stave or carpet-like rupture. We observed inactivation of the channeling activity when C70 was encapsulated inside the cage, indicating that ions and small molecules are transported through the cages' cavity. Furthermore, we have demonstrated that ion transport selectivity can be modulated by altering the electronic environment of the cavity through metalation of the porphyrins. Overall, our study introduces a new class of unimolecular artificial channels, achievable through alkyne metathesis, that form shape-persistent covalent organic molecular cages with isolated accessible internal cavities. This work could also guide the design of novel artificial transmembrane channel molecules with promising applications in biological systems.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Brandon Bishop: Writing – review & editing, Writing – original draft, Investigation, Formal analysis, Data curation. Shaofeng Huang: Writing – review & editing, Writing – original draft, Investigation, Formal analysis, Data curation. Hongxuan Chen: Data curation. Haijia Yu: Investigation, Data curation. Hai Long: Investigation, Data curation. Jingshi Shen: Supervision. Wei Zhang: Writing – review & editing, Writing – original draft, Supervision, Project administration, Formal analysis, Conceptualization.
We thank E. Dempsey (Colorado School of Mines) for the assistance with MALDI measurement, and B. Hohman (University of Colorado) for the help with DLS test. S. H. thanks the partial financial support by the Summer Graduate School Fellowship from University of Colorado Boulder.
Supplementary material associated with this article can be found, in the online version, at doi:
E. Neher, Angew. Chem. Int. Ed. 31 (1992) 824–829. doi: 10.1002/anie.199208241
A.I. Cooper, Angew. Chem. Int. Ed. 50 (2011) 996–998. doi: 10.1002/anie.201006664
P. Agre, Angew. Chem. Int. Ed. 43 (2004) 4278–4290. doi: 10.1002/anie.200460804
R. McCaffrey, H. Long, Y. Jin, et al., J. Am. Chem. Soc. 136 (2014) 1782–1785. doi: 10.1021/ja412606t
X.B. Hu, Z. Chen, G. Tang, J.L. Hou, Z.T. Li, J. Am. Chem. Soc. 134 (2012) 8384–8387. doi: 10.1021/ja302292c
J.R. Werber, C.O. Osuji, M. Elimelech, Nat. Rev. Mater. 1 (2016) 16018. doi: 10.1038/natrevmats.2016.18
D. Xia, P. Wang, X. Ji, et al., Chem. Rev. 120 (2020) 6070–6123. doi: 10.1021/acs.chemrev.9b00839
N. Busschaert, P.A. Gale, Angew. Chem. Int. Ed. 52 (2013) 1374–1382. doi: 10.1002/anie.201207535
Y.J. Jeon, H. Kim, S. Jon, et al., J. Am. Chem. Soc. 126 (2004) 15944–15945. doi: 10.1021/ja044748j
J.L. Atwood, L.J. Barbour, A. Jerga, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 4837–4841. doi: 10.1073/pnas.082659799
Z. Liu, S.K.M. Nalluri, J.F. Stoddart, Chem. Soc. Rev. 46 (2017) 2459–2478. doi: 10.1039/C7CS00185A
Y. Zhou, K. Jie, R. Zhao, F. Huang, Adv. Mater. 32 (2020) 1904824. doi: 10.1002/adma.201904824
R. Liu, C. Luo, Z. Pang, et al., Chin. Chem. Lett. 34 (2023) 107518. doi: 10.1016/j.cclet.2022.05.032
X. Wu, Luke W. Judd, Ethan N.W. Howe, et al., Chem 1 (2016) 127–146. doi: 10.1016/j.chempr.2016.04.002
T.G. Johnson, A. Sadeghi-Kelishadi, M.J. Langton, J. Am. Chem. Soc. 144 (2022) 10455–10461. doi: 10.1021/jacs.2c02612
A. Mondal, S.N. Save, S. Sarkar, et al., J. Am. Chem. Soc. 145 (2023) 9737–9745. doi: 10.1021/jacs.3c01451
J. Shen, D. R, Z. Li, et al., Angew. Chem. Int. Ed. 62 (2023) e202305623. doi: 10.1002/anie.202305623
S. Negin, M.M. Daschbach, O.V. Kulikov, N. Rath, G.W. Gokel, J. Am. Chem. Soc. 133 (2011) 3234–3237. doi: 10.1021/ja1085645
O.V. Kulikov, R. Li, G.W. Gokel, Angew. Chem. Int. Ed. 48 (2009) 375–377. doi: 10.1002/anie.200804099
B. Gong, Z. Shao, Acc. Chem. Res. 46 (2013) 2856–2866. doi: 10.1021/ar400030e
A.J. Helsel, A.L. Brown, K. Yamato, et al., J. Am. Chem. Soc. 130 (2008) 15784–15785. doi: 10.1021/ja807078y
A. Cataldo, K. Norvaisa, L. Halgreen, et al., J. Am. Chem. Soc. 145 (2023) 16310–16314. doi: 10.1021/jacs.3c04631
Y. Itoh, S. Chen, R. Hirahara, et al., Science 376 (2022) 738–743. doi: 10.1126/science.abd0966
C.Y. Wu, Y.N. Gao, Z.H. Zhang, et al., Chin. Chem. Lett. 35 (2024) 109649. doi: 10.1016/j.cclet.2024.109649
H. Ma, R. Ye, L. Jin, et al., Chin. Chem. Lett. 34 (2023) 108355. doi: 10.1016/j.cclet.2023.108355
J.Y. Chen, Q. Xiao, H. Behera, L. Hou, Chin. Chem. Lett. 31 (2020) 77–80. doi: 10.1016/j.cclet.2019.05.009
S. Zhang, I. Boussouar, H. Li, Chin. Chem. Lett. 32 (2021) 642–648. doi: 10.1016/j.cclet.2020.06.035
M. Jung, H. Kim, K. Baek, K. Kim, Angew. Chem. Int. Ed. 47 (2008) 5755–5757. doi: 10.1002/anie.200802240
N. Sakai, S. Matile, Angew. Chem. Int. Ed. 47 (2008) 9603–9607. doi: 10.1002/anie.200803300
U. Devi, J.R.D. Brown, A. Almond, S.J. Webb, Langmuir 27 (2011) 1448–1456. doi: 10.1021/la104152s
A. Razmjou, M. Asadnia, E. Hosseini, A. Habibnejad Korayem, V. Chen, Nat. Commun. 10 (2019) 5793. doi: 10.1038/s41467-019-13648-7
P. Xin, P. Zhu, P. Su, J.L. Hou, Z.T. Li, J. Am. Chem. Soc. 136 (2014) 13078–13081. doi: 10.1021/ja503376s
C. Tribet, F. Vial, Soft Matter 4 (2008) 68–81. doi: 10.1039/B708431P
T. Jiang, A. Hall, M. Eres, et al., Nature 577 (2020) 216–220. doi: 10.1038/s41586-019-1881-0
T. Yan, S. Liu, C. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202210214. doi: 10.1002/anie.202210214
L. Yu, M. Wang, X. Li, X. Hou, Chin. Chem. Lett. 34 (2023) 107785. doi: 10.1016/j.cclet.2022.107785
J. Hao, W. Wang, J. Zhao, et al., Chin. Chem. Lett. 33 (2022) 2291–2300. doi: 10.1016/j.cclet.2021.10.011
C. Ma, Y. Zhang, Z. Jiao, et al., Chin. Chem. Lett. 31 (2020) 1635–1639. doi: 10.1016/j.cclet.2019.09.010
B. Baumeister, N. Sakai, S. Matile, Angew. Chem. Int. Ed. 39 (2000) 1955–1958. doi: 10.1002/1521-3773(20000602)39:11<1955::AID-ANIE1955>3.0.CO;2-M
V. Percec, A.E. Dulcey, V.S.K. Balagurusamy, et al., Nature 430 (2004) 764–768. doi: 10.1038/nature02770
M.R. Ghadiri, J.R. Granja, L.K. Buehler, Nature 369 (1994) 301–304. doi: 10.1038/369301a0
R. Chapman, M. Danial, M.L. Koh, K.A. Jolliffe, S. Perrier, Chem. Soc. Rev. 41 (2012) 6023–6041. doi: 10.1039/c2cs35172b
L. Yuan, P. Jiang, J. Hu, et al., Chin. Chem. Lett. 33 (2022) 2026–2030. doi: 10.1016/j.cclet.2021.09.089
J. Shen, R. Ye, Z. Liu, H. Zeng, Angew. Chem. Int. Ed. 61 (2022) e202200259. doi: 10.1002/anie.202200259
A. Roy, J. Shen, H. Joshi, et al., Nat. Nanotechnol. 16 (2021) 911–917. doi: 10.1038/s41565-021-00915-2
S. Qi, C. Zhang, H. Yu, et al., J. Am. Chem. Soc. 143 (2021) 3284–3288. doi: 10.1021/jacs.0c12128
F. Chen, J. Shen, N. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 1440. doi: 10.1002/anie.201906341
J. Shen, C. Ren, H. Zeng, Acc. Chem. Res. 55 (2022) 1148–1159. doi: 10.1021/acs.accounts.1c00804
C. Wang, S. Wang, H. Yang, et al., Angew. Chem. Int. Ed. 60 (2021) 14836. doi: 10.1002/anie.202102838
C. Ren, F. Chen, R. Ye, et al., Angew. Chem. Int. Ed. 58 (2019) 8034. doi: 10.1002/anie.201901833
S.C. Balmert, S.R. Little, Adv. Mater. 24 (2012) 3757–3778. doi: 10.1002/adma.201200224
A. Barba-Bon, G. Salluce, I. Lostalé-Seijo, et al., Nature 603 (2022) 637–642. doi: 10.1038/s41586-022-04413-w
J. Tian, P.K. Thallapally, B.P. McGrail, CrystEngComm 14 (2012) 1909–1919. doi: 10.1039/c2ce06457j
R.C. Huxford, J. Della Rocca, W. Lin, Curr. Opin. Chem. Biol. 14 (2010) 262–268. doi: 10.1016/j.cbpa.2009.12.012
T. Tozawa, J.T.A. Jones, S.I. Swamy, et al., Nat. Mater. 8 (2009) 973–978. doi: 10.1038/nmat2545
S. Huang, Z. Lei, Y. Jin, W. Zhang, Chem. Sci. 12 (2021) 9591–9606. doi: 10.1039/D1SC01881G
G. Zhang, M. Mastalerz, Chem. Soc. Rev. 43 (2014) 1934–1947. doi: 10.1039/C3CS60358J
C. Zhang, Q. Wang, H. Long, W. Zhang, J. Am. Chem. Soc. 133 (2011) 20995–21001. doi: 10.1021/ja210418t
Q. Wang, C. Yu, C. Zhang, et al., Chem. Sci. 7 (2016) 3370–3376. doi: 10.1039/C5SC04977F
Y. Jin, B.A. Voss, R.D. Noble, W. Zhang, Angew. Chem. Int. Ed. 49 (2010) 6348–6351. doi: 10.1002/anie.201001517
M. Liu, L. Zhang, M.A. Little, et al., Science 366 (2019) 613–620. doi: 10.1126/science.aax7427
Y. Jin, B.A. Voss, A. Jin, et al., J. Am. Chem. Soc. 133 (2011) 6650–6658. doi: 10.1021/ja110846c
R. Afrasiabi, H.B. Kraatz, Chem. Eur. J. 19 (2013) 15862–15871. doi: 10.1002/chem.201303116
T. Mitra, K.E. Jelfs, M. Schmidtmann, et al., Nat. Chem. 5 (2013) 276–281. doi: 10.1038/nchem.1550
P.T. Smith, B.P. Benke, Z. Cao, et al., Angew. Chem. Int. Ed. 57 (2018) 9684–9688. doi: 10.1002/anie.201803873
L. An, M.R. Narouz, P.T. Smith, P. De La Torre, C.J. Chang, Angew. Chem. Int. Ed. 62 (2023) e202305719. doi: 10.1002/anie.202305719
Y. Hu, S. Huang, L.J. Wayment, et al., Cell Rep. Phys. Sci. 4 (2023) 101285. doi: 10.1016/j.xcrp.2023.101285
N. Sun, C. Wang, H. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 18011–18016. doi: 10.1002/anie.201908703
L. Qiu, R. McCaffrey, Y. Jin, et al., Chem. Sci. 9 (2018) 676–680. doi: 10.1039/C7SC03148C
B.P. Benke, P. Aich, Y. Kim, et al., J. Am. Chem. Soc. 139 (2017) 7432–7435. doi: 10.1021/jacs.7b02708
H.G. Lee, A. Dhamija, C.K. Das, et al., Angew. Chem. Int. Ed. 62 (2023) e202214326. doi: 10.1002/anie.202214326
Q. Wang, C. Zhang, B.C. Noll, et al., Angew. Chem. Int. Ed. 53 (2014) 10663–10667. doi: 10.1002/anie.201404880
Q. Wang, C. Yu, H. Long, et al., Angew. Chem. Int. Ed. 54 (2015) 7550–7554. doi: 10.1002/anie.201501679
X. Yang, S. Huang, M. Ortiz, et al., Org. Chem. Front. 8 (2021) 4723–4729. doi: 10.1039/D1QO00685A
Y. Du, H. Yang, C. Zhu, et al., Chem. Eur. J. 22 (2016) 7959–7963. doi: 10.1002/chem.201505174
M. Doktorova, F.A. Heberle, B. Eicher, et al., Nat. Protoc. 13 (2018) 2086–2101. doi: 10.1038/s41596-018-0033-6
S. Qi, J. Tian, J. Zhang, et al., CCS Chem. 4 (2022) 1850–1857. doi: 10.31635/ccschem.021.202101144
A. Gilles, M. Barboiu, J. Am. Chem. Soc. 138 (2016) 426–432. doi: 10.1021/jacs.5b11743
X. Wu, P.A. Gale, Chem. Commun. 57 (2021) 3979–3982. doi: 10.1039/D1CC01038G
W.H. Chen, X.B. Shao, S.L. Regen, J. Am. Chem. Soc. 127 (2005) 12727–12735. doi: 10.1021/ja053527q
W. SzymaŃski, D. Yilmaz, A. Koçer, B.L. Feringa, Acc. Chem. Res. 46 (2013) 2910–2923. doi: 10.1021/ar4000357
S. Litvinchuk, H. Tanaka, T. Miyatake, et al., Nat. Mater. 6 (2007) 576–580. doi: 10.1038/nmat1933
M. Tagliazucchi, I. Szleifer, Mater. Today 18 (2015) 131–142. doi: 10.1016/j.mattod.2014.10.020
T. Muraoka, T. Endo, K.V. Tabata, et al., J. Am. Chem. Soc. 136 (2014) 15584–15595. doi: 10.1021/ja5070312
D.A. Edwards, M.R. Prausnitz, R. Langer, J.C. Weaver, J. Control. Release 34 (1995) 211. doi: 10.1016/0168-3659(94)00132-E

Scheme 1 Synthesis of molecular cages. Conditions: (a) Mo catalyst, CHCl3, 5 Å molecular sieves, reflux; (b) Zn(OAc)2, CHCl3/MeOH, reflux; (c) KOH, THF/MeOH/H2O, reflux.

Figure 1 (A) Schematic representation of the ion transport experiment using LUVs loaded with the pH-sensitive fluorophore HPTS and exposed to a pH gradient (inside pH 7.0, outside pH 7.6). (B) Normalized (Norm.) HPTS fluorescence traces as a function of time in the presence of various concentrations of C1. Extravesicular buffer solution: 10 mmol/L HEPES, 100 mmol/L NaCl, pH 7.6; intravesicular buffer solution: 10 mmol/L HEPES, ~0.1 mmol/L HPTS, 100 mmol/L NaCl, pH 7.0. Triton X-100 (16 µL, 20% v/v) was added at 480 s to achieve complete disruption of LUVs. (C) Norm. HPTS fluorescence traces as a function of time in the presence or absence of C1 (20 µmol/L). Extravesicular buffer solution: 10 mmol/L HEPES, 100 mmol/L MX, pH 7.6; intravesicular buffer solution: 10 mmol/L HEPES, ~0.1 mmol/L HPTS, 100 mmol/L NaX, pH 7.0. Triton X-100 (16 µL, 20%, v/v) was added at 480 s to achieve complete disruption of LUVs. (D) Linear correlation between the observed ion transport rate and the concentration of C1.

Figure 2 (A) Norm. HPTS fluorescence traces as a function of time in the presence or absence of C1 (20 µmol/L). Extravesicular buffer solution: 10 mmol/L HEPES, 100 mmol/L MCl, pH 7.6; intravesicular buffer solution: 10 mmol/L HEPES, ~0.1 mmol/L HPTS, 100 mmol/L NaCl, pH 7.0. Triton X-100 (16 µL, 20%, v/v) was added at 480 s to achieve complete disruption of LUVs. V stands for valinomycin. (B) Norm. HPTS fluorescence traces as a function of time in the presence or absence of C1 (20 µmol/L). Extravesicular buffer solution: 10 mmol/L HEPES, 100 mmol/L MCl, pH 7.6; intravesicular buffer solution: 10 mmol/L HEPES, ~0.1 mmol/L HPTS, 100 mmol/L NaCl, pH 7.0. Triton X-100 (16 µL, 20% v/v) was added at 480 s to achieve complete disruption of LUVs. (C) Snapshot from a 10 ns molecular dynamics simulation. The cage is shown in blue and lipids are in light green. Oxygen atoms of water molecules are in red and hydrogen atoms are in white. (D) Normalized HPTS fluorescence traces as a function of time in the presence of C1 (light blue), Zn-C1 (blue), C2 (lightest green), C3 (green), C4 (light green), C5 (dark green), and C1-C70 (dark blue). The concentrations of the cages in total lipids in (D) were 20 µmol/L (1.50 mol% relative to lipid).

Figure 3 (A) Normalized fluorescence traces from the calcein transport assay for various concentrations of C1. (B) Normalized fluorescence traces from the calcein transport assay in the presence of C1 (light green), Zn-C1 (lightest green), C2 (green), C4 (Dark green). (C) Fluorescence image of HEK 293 T cells after the exposure to calcein in the presence of C1. (D) Fluorescence image of HEK 293 T cells after the exposure to calcein in absence of C1.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: