MOE Key Laboratory of Macromolecular Synthesis and Functionalization, International Research Center for X Polymers, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
Received Date:
17 February 2024 Accepted Date:
15 April 2024 Revised Date:
14 April 2024 Available Online:
15 November 2024
Abstract:
In 2023, The MOE Key Laboratory of Macromolecular Synthesis and Functionalization in Zhejiang University had achieved several important results in the five research directions. First, for controllable catalytic polymerization, a new silicon-centered organoboron binary catalyst was developed for copolymerization of epoxides, and a series of cooperative organocatalysts were proposed for ring-opening copolymerization of chalcogen-rich monomers. Second, with respect to microstructure and rheology, axially encoded metafiber demonstrated its capacity for integrating multiple electronics, while artificial nacre materials showed improved strength and toughness due to interlayer entanglement. Third, concerning separating functional polymers, interfacial polymerization was monitored via aggregation-induced emission, and vacuum filtration was applied to assist interfacial polymerization. Fourth, in terms of biomedical functional polymers, we designed antibacterial materials such as a novel quaternary ammonium salt that enables polyethylene terephthalate recycling and its antibacterial function, nanozyme-armed phage proved its efficiency in combating bacterial infection, and also transition metal nanoparticles showed capacities in antibacterial treatments. We also made achievements in biomedical materials, including polymeric microneedles for minimally invasive implantation and functionalization of cardiac patches, as well as ROS-responsive/scavenging prodrug/miRNA balloon coating to promote drug delivery efficiency. Besides, methods and mechanisms of RNA labeling has been developed. Fifth, about photo-electro-magnetic functional polymers, through-space conjugation was successfully manipulated by altering subunit packing modes, room-temperature phosphorescent hydrogels were synthesized via polymerization-induced crystallization of dopant molecules, and single crystals of both fullerene and non-fullerene acceptors were grown in crystallized organogel, with their photodetection performance further explored. The related works are reviewed in this paper.
The MOE Key Laboratory of Macromolecular Synthesis and Functionalization was founded in Zhejiang University in 2005, with a primary emphasis on both fundamental and applied polymer science. The laboratory established five research directions, which encompass controllable catalytic polymerization, microstructure and rheology, photo-electro-magnetic functional polymers, biomedical functional polymers, and separating functional polymers. Focusing on the frontiers of polymer science advancements, the laboratory has made breakthroughs in three main areas. Firstly, it synthesizes novel functional polymers with controllable structures by means via a deeper exploration of methods and mechanisms for controlled polymerization. Secondly, it investigates the structure-property relationship through the lens of microstructure and rheology, aimed at elucidating the formation and evolution of polymer morphology during fabrication and its consequential effects on material properties. Thirdly, it explores the formulation of novel strategies for crafting polymer-based functional materials, to attain high-performance polymer materials or devices across diverse application domains [1–3].
In 2023, the laboratory generated abundant research achievements across the aforementioned five research domains (Fig. 1). Thus, this manuscript has been prepared to evaluate a selection of pioneering studies published in 2023, encompassing all five research directions. In controllable catalytic polymerization, we explore metal-free organoboron catalysts and organocatalysis for ring-opening copolymerization. In microstructure and rheology, we design axially encoded metafibers and artificial nacre materials. In separating functional polymers, we monitor interfacial polymerization. In biomedical functional polymers, we obtain antibacterial materials such as antibacterial polyesters, microenvironment-activated nanozyme-armed bacteriophages and antibacterial nanomaterials, and also biomedical materials such as polymer cardiac patches and drug coating, as well as attempts in RNA labelling. In photo-electro-magnetic functional polymers, we investigate multiaryl alkanes through-space conjugation manipulation, room-temperature phosphorescent hydrogels and long-range ordered bulk-heterojunctions.
Figure 1
Figure 1.
Schematic illustration about the key progresses of MOE Key Laboratory of Macromolecular Synthesis and Functionalization in 2023. Adapted with permission [5,44,48,105,118,121,122,130,151,183]. Copyright 2023, Wiley-VCH; Copyright 2023, Elsevier; Copyright 2023, American Chemical Society; Copyright 2023, Springer Nature; Copyright 2024, Royal Society of Chemistry; Copyright 2023, National Natural Science Foundation of China.
First, we develop a novel organoboron catalyst. Inspired by the synergistic effect between two boron centers in bicomponent catalyst system, in 2020, Wu’s group designed a series of dinuclear bifunctional catalysts for the ring-opening polymerization of epoxides, where boron and ammonium salt were covalently connected within a single molecule (Fig. 2, blue) [4]. In 2023, Wu’s group reported a new silicon-centered binary catalyst (Fig. 2, red) with higher activity than its bifunctional analogue (blue) [5]. They replaced ammonium salts with a silicon atom, and the prepared the binary silicon-centered organoboron catalyst exhibited 15 times the activity of its bifunctional ammonium salts-centered analogue at a low feed [catalyst]/propylene oxide molar ratio of 50,000/1. Mechanism studies revealed that the central silicon atom of the catalyst possessed a certain degree of electron deficiency, which enabled its involvement in the co-activation of monomers and the delivery of growing polymer chains. And such findings might broaden the future design of intramolecular organoboron catalysts.
Figure 2
Figure 2.
Silicon-centered binary (red) catalyst with higher activity than its bifunctional analogue (blue) for ring-opening polymerization of propylene oxide proposed by Wu et al. Reproduced with permission [5]. Copyright 2023, Wiley-VCH.
Second, we explore cooperative organocatalysis for ring-opening copolymerization of chalcogen-rich monomers. Ring-opening (co)polymerization (RO(CO)P) has been determined as a versatile method toward synthesizing a wide range of chalcogen-rich polymers [6]. The synthetic route features favorable characteristics of atom economical, mild reaction conditions, high and controlled molecular weight, etc. [7,8]. The development of efficient and selective catalysts is one of the frontiers of this area [9]. Organocatalysis has provided a plenty of opportunities for polymer synthesis, which led to the conceptualization of “organocatalytic polymerization” in the early 21st century [10–12]. Especially, the organocatalytic RO(CO)P has blossomed greatly in the past 20 years [13]. The family of the reported organocatalysts includes Brønted/Lewis acids, Brønted/Lewis bases, and bifunctional catalyst systems [14]. The advancement of robust, versatile, and efficient organocatalysts for metal-free RO(CO)P remains challenging.
The ROCOP of CO2 and propylene oxide (PO) is a facile strategy to yield biodegradable poly(propylene carbonate) (PPC) with excellent oxygen barrier properties [15–17]. Alkyl borane-based Lewis pairs have been determined as effective catalysts for the ROCOP [18–20]. Zhang and co-workers employed the frustrated Lewis pair (FLP) consisting of tertiary phosphine and trialkylborane as the metal-free catalyst system for the ROCOP of PO and CO2 (Fig. 3a) [21]. The reasonable matching of the electronic properties and steric hindrances of Lewis pairs has a huge impact on the activity and selectivity of the copolymerization. The Lewis base of PtBu3 is highly nucleophilic and has a large steric hindrance. The Lewis acid of BnBu3 has a medium Lewis acidity and a medium steric hindrance. The FLP consisting of PtBu3 and BnBu3 exhibited a moderate activity (with TOF of up to 447 h−1) and a high initiation efficiency toward the ROCOP of CO2 and PO. The obtained PPCs possessed well-defined structure with controlled molecular weights of 8.0–19.9 kDa, narrow dispersities of < 1.1, and high carbonate contents of 96–99 mol%. The excellent catalytic performance of the Lewis pair confirms a favorable catalytic methodology for CO2 and PO copolymerization.
Figure 3
Figure 3.
(a) ROCOP of PO and CO2 using BnBu3 combined with PtBu3. Reproduced with permission [21]. Copyright 2023, American Chemical Society. (b) ROCOP of TMO and CS2 using TEB combined with PPNCl. Reproduced with permission [25]. Copyright 2023, American Chemical Society. (c) ROCOP of COS, PO, and rac-lactide to yield stereo- and sequence-defined copolymers using various multifunctional organocatalysts. Reproduced with permission [29]. Copyright 2023, American Chemical Society.
CS2 is a low-cost chemical and is always discarded as a pollutant from industries [22]. The transformation of CS2 into useful sulfur-containing polymers is of great significance to the environment and polymer science [23]. However, the ROCOP of CS2 and cyclic ethers always suffers from complicated polymer structure and massive small-molecular cyclic thiocarbonate by-products [24]. Zhang and co-workers used the bicomponent organocatalyst system consisting of an alkyl borane (triethylboron, TEB) and an onium salt (bis(triphenylphosphine)iminium chloride, PPNCl) for the ROCOP of CS2 and trimethylene oxide (TMO) (Fig. 3b) [25]. With the organocatalyst system, the ROCOP is highly efficient with a TOF of up to 1103 h−1, avoids by-products, and yields unprecedented polythiocarbonates with complete TMO-alt-CS2 regular units. The obtained well-defined polythiocarbonate demonstrates a melting temperature of 83 ℃, ultimate tensile strength of 20.7 ± 3.1 MPa, and elongation at a break of 480% ± 20%. The copolymerization mechanism was clearly demonstrated by DFT calculations: TEB activates TMO and generates a borate complex with the growing anion.
COS is also a low-cost monomer for the production of sulfur-containing polymers, which is produced industrially from CO and elemental sulfur [26]. The ROCOP of PO and COS was firstly reported by Zhang and co-workers in 2013 [27]. The ROCOP has been demonstrated as a facile route to yield polythiocarbonates with favorable optical and mechanical properties [28]. Recently, Zhang and co-workers report a one-pot and switchable copolymerization of COS, PO, and rac-lactide, affording various stereo- and sequence-defined copolymers bearing isotactic polylactide and alternating polythiocarbonate blocks (Fig. 3c) [29]. For the copolymerization, they developed numerous multifunctional organocatalysts incorporating boron and Takemoto’s catalyst in one molecular. The site-monomer specific recognition mechanism was proposed: The boron site activates PO through B-O bonds; the thiourea site activates lactide through hydrogen bonds; and the amine site activates initiator/chain end. The obtained block copolymers exhibit high toughness and ductility: P10 possesses a molecular weight of 92.3 kDa and exhibits ultimate tensile strength of 59 ± 3 MPa and elongation at break of 222% ± 25%. This strategy provides unique insights into the design of efficient organocatalysts and valuable sustainable polymers.
3.
Microstructure and rheology
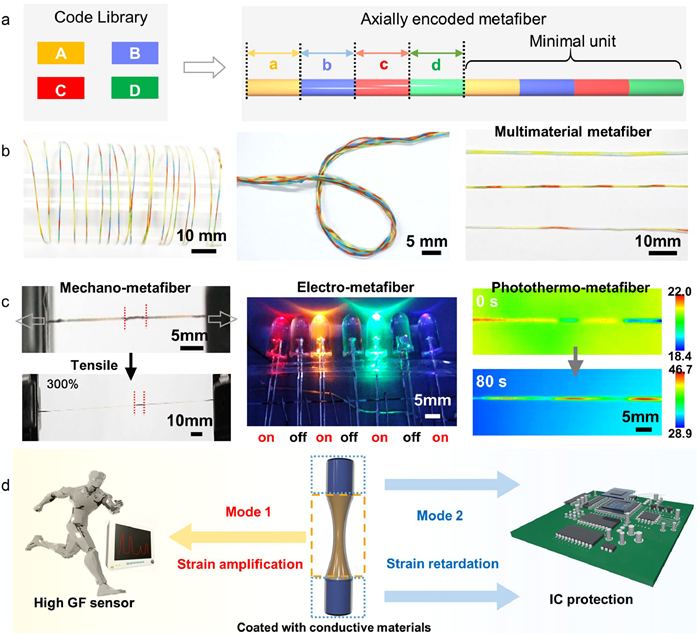
First, we design axially encoded metafibers for fiber electronics. Fibers are fundamental building blocks of ubiquitous textiles and organisms and have rapidly evolved to carry complex functions and information [30–33]. Beyond conventional homogeneous fibers with homogenous constitutions, fiber with meta-structure, named as metafiber, can integrate multiple customized meta-structures, such as electrodes and electrolytes for the fabrication of fiber electronics [34]. The design of metafiber requires the encoding of heterogeneous compositions along the radial or axial directions [35]. Theoretically, the axial meta-structures can render metafiber high integration density by fully utilizing the ultralong one-dimensional profile, which should realize the highest integration density and provide vast space for designing smart textiles. To date, integrating massive customized meta-structures along the fiber axial direction still remains a challenge. We proposed a general wet-spinning method to prepared axial metafibers by microfluidic sequence spinning (MSS) system and extended the new metafiber in sensors and electronic textiles [36,37].
We built the MSS system to fabricate axially encoded metafibers with customized meta-structural units (Fig. 4a) [36]. The material composition distribution along the fiber axial direction was described by a well-defined mathematical model, which was further converted to the sequence spinning program. Through shifting the flow of diverse spinning solution, the spinning dopes were converted into designed sequential arrangement. Notably, the code length and arrangement can be precisely controlled by tailoring the computer program. The axially integrated sequential codes were extended to various species including color, morphology, material compositions and functions, which greatly expands the fiber family (Fig. 4b).
Figure 4
Figure 4.
(a) Designing concept of axially encoded metafiber from the code library. (b) Images of axially encoded metafibers. (c) Metafibers with axially functional asymmetry. Reproduced with permission [36]. Copyright 2023, Elsevier. (d) Schematic illustration of the localized strain engineering for highly sensitive sensor and IC protection. Reproduced with permission [37]. Copyright 2023, Wiley-VCH.
We revealed that the encoded heterogeneity breaks the fiber axial symmetry, converting it to a type of one-dimensional metamaterial assembled by multiple functional sequences with independent information and properties (Fig. 4c). For instance, we found that mechano-metafibers composed of alternatively encoded soft and stiff polyurethane sequences exhibit localized mechanical properties [37]. Under homogeneous tensile loading, the strain was greatly expressed in the soft sequences but highly retarded in the stiff sequences, and the strain amplification/retardation ratio was tailored by adjusting the content of the stiff sequences (Fig. 4d). The unique nonlinear deformation behavior was ideal in fiber electronics for localized strain engineering, enabling the design of highly sensitive sensors, stretchable electronics, high-voltage output supercapacitors and axial electroluminescent arrays. Axially encoded metafiber brings insights into designing fibrous metamaterials, and provides a route for integrating multiple electronics in one single fiber.
Second, we toughen and strengthen artificial nacre materials through strong interlayer entanglement. Biological materials, such as the shell of mollusks, wood, bones, and spider fibers, usually behave strong and tough to adapt to their complex mechanical functions [38,39]. The underlying spirit to reconcile the conflict between strength and toughness is the powerful dissipative mechanisms that express through their multiscale hierarchical structures. The understanding of natural nacre has inspired the design of rich nacre-inspired materials that possess high strength and toughness simultaneously. However, to fully harness the powerful dissipative spirit of natural materials remains a grand challenge.
Chain entanglement is a physical interlocking characteristic of polymer chains and the topology constraint mechanism of entangle molecular networks contribute the high strength and toughness of synthetic polymeric materials. To utilize this mechanism, we reported a new interlayer dissipation mechanism by introducing strong interchain entanglement of ultra-high molecular weight polymer, serving as folded proteins in the natural nacre (Fig. 5a) [40]. We prepared three kinds of nacre-mimic materials with strong entanglement, named as entangled nacres, extending from nacre fibers, films to papers (Fig. 5b). These entangled nacre materials exhibited a breakthrough combination of strength and toughness. We revealed that the simultaneous increase of strength and toughness follows the enhancing chain entanglement and increasing molecular weight (Fig. 5c). The enhancing entanglement uncovers a new artificial mechanism to harness the spirit of evolutional biological materials. This mechanism can be facilely extended to a broad kind of materials, including other 2D sheets and nanoplatelets, and opens an avenue for developing biomimetic materials with the integration of high structural properties and excellent functional applications.
Figure 5
Figure 5.
Structural model and overall properties of entangled nacre materials. (a) Failure models of entangled and regular nacre materials. Highly entangled polymer chains (crossing loops labelled in circles) strengthen energy dissipation to prevent catastrophic separation of sliding bricks than ordinary weak entangled case. (b) Photos of entangled nacre materials, including molecular entangled nacre fibers and films and nano entangled nacre papers. (c) The simultaneous increasing trend of toughness and strength of entangled nacres as Mw increases. Copied with permission [40]. Copyright 2023, American Chemical Society.
Polyamide nanofiltration and reverse osmosis membranes are extensively employed in diverse industrial processes that necessitate separation. These membranes are typically manufactured using interfacial polymerization. Monitoring the microscale dynamics of interfacial polymerization remains a challenge, yet it is crucial for understanding and subsequently regulating this process. We have developed a novel in-situ technique that enables real-time monitoring of polymer growth during interfacial polymerization between amine and acryl chloride monomers using aggregation-induced emission (AIE) [41]. A fluorescent monomer, tetrakis(4-aminophenyl) ethylene (TAPE), was applied to track the monomer consumption during this process by detecting the fluorescence intensity in the monomer solution, the variation of which reflected the reaction-limited and diffusion limited stages. This year, we expanded this technique to other interfacial polymerization systems (Fig. 6a) [42]. TAPE and toluene diisocyanate (TDI) were selected as the monomers to synthesize polyurea nanofilms at the interface. We selected TAPE and toluene diisocyanate (TDI) as the monomers to synthesize polyurea nanofilms at the interface. In comparison to the previous system, the polymer formed at the interface exhibited a strong AIE effect, enabling the visualization of the interface polymerization process under a laser scanning confocal microscope. The current method has a limitation due to the poor solubility of the AIE molecule, TAPE in water, requiring the use of an ionic liquid to replace the aqueous phase, which differs from the conventional water-alkaline interface.
Figure 6
Figure 6.
(a) Schematic illustration of the in situ and real-time monitoring of the interfacial polymerization based on the AIE effect. Reproduced with permission [42]. Copyright 2023, American Chemical Society. (b) Schematic of the effects of different alkane solvents on interfacial polymerization. Reproduced with permission [45]. Copyright 2023, Elsevier. (c) Schematic illustration of blade-coating-spraying-interfacial polymerization. Reproduced with permission [46]. Copyright 2023, Elsevier. (d) Schematic illustration of vacuum-assisted interfacial polymerization. Reproduced with permission [47]. Copyright 2023, Elsevier.
In addition to developing methods to investigate the mechanisms of interfacial polymerization, we also focus on the industrial interfacial polymerization process and its regulation methods. Our previous work involved the development of a series of strategies, including the applying interlayers, engineering substrate surfaces, and designing reaction systems [43,44]. In 2023, we began focusing on facile strategies with potential applications in industrial production. For instance, we explored the effects of using isoalkanes, as opposed to the more commonly used hexane in laboratory, in interfacial polymerization. Isoalkanes are favored in industrial production due to their relatively low volatility, ensuring safer production environments. Due to their higher viscosity and increased interfacial tension, isoalkanes not only contribute to a thinner reaction interface but also restrict the diffusion of acyl chlorides, enabling controlled interfacial polymerization (Fig. 6b) [45]. Due to the relatively low volatility of isoalkanes, the future industrial production could involve the feasible spraying coating of the organic phase. We investigated a sequential process involving blade coating, spraying, and interfacial polymerization for the scalable preparation of polyamide nanofilms with high desalination performance (Fig. 6c) [46]. The resulting polyamide membranes exhibit a permeance flux of 1.89 L m−2 h−1 bar−1 with 97.4% NaCl rejection, and this method can save nearly 99% of organic solvent compared to conventional ones. In addition to these efforts, we have also explored novel processes for conducting interfacial polymerization. We discovered that vacuum-assisted interfacial polymerization (Fig. 6d) could expand the options for substrate materials, widen the monomer concentration window, and improve the stability and reproducibility of nanofiltration performance in interfacially polymerized polyamide membranes [47]. Vacuum filtration of the aqueous monomer solution enables a uniform distribution of monomers on the substrate compared to the conventional drying process. Additionally, we have developed a transfer-printing compositing strategy to mitigate the effects of substrates on interfacial polymerization [48].
5.
Biomedical functional polymers
5.1
Antibacterial materials
First, we design antibacterial polyesters via conventional polycondensation. Polyethylene terephthalate (PET), renowned for its cost-effectiveness, durability, and robust stability, stands as the predominant plastic in the realms of textiles and bottle production, with a global annual output nearing 70 million tons [49–53]. As the global demand for PET has experienced significant growth, the recycling of PET waste from plastic bottles and textiles presents an effective means to alleviate environmental pressures stemming from plastic pollution [54–58]. Presently, PET recycling predominantly relies on mechanical recycling and chemical solvolysis. Although the recovery rate through physical recycling is increasing, most recycled PET is converted into low-value products, thereby constraining the extensive utilization of physical recycling, primarily due to a lack of economic incentives for businesses. Chemical recycling, an alternative method, converts polymers into small-molecule chemical products, such as dimethyl terephthalate, etc. However, due to cumbersome purification steps, heavy use of organic solvents, and high energy consumption, the resulting monomer becomes more expensive than the original monomer derived from petroleum, constraining the applicability of chemical recycling. The most promising approach involves directly upcycling PET into high-value materials to offset recycling costs. Notably, the high-value of antibacterial materials, coupled with a burgeoning market demand, has spurred extensive research and application in this domain. Concurrently, public health concerns related to bacterial infections are propelling researchers to develop new techniques for preparing antibacterial materials. Directly converting waste PET into antibacterial PET holds significant promise.
The potent antibacterial activities of quaternary ammonium salts (QACs) make them compelling candidates for the production of antibacterial PET. However, classical QACs are hindered by low thermostability. The challenge lies in enhancing the thermal stability of existing antibacterial monomers, given that melt polycondensation or solid-state reaction (SSR) of PET often require high temperatures (> 200 ℃) [2,59-63]. Our previous research has demonstrated that integrating a diaromatic ring structure into QACs can significantly enhance their thermal stability. For instance, [C16][SIPA], which incorporates an imidazole and a benzene ring, exhibits remarkable thermal stability with a decomposition temperature of 361 ℃ [64]. And then, we have reported a convenient and environmentally friendly method for the large-scale synthesis of [C16][SIPA], achieved through the reaction of readily available QACs and NaSIPA in deionized water. Building upon [C16][SIPA], we have successfully synthesized antibacterial poly(ethylene succinate) via melt polycondensation [64].
For recycling waste PET, we have developed a novel, cost-effective reaction route based on SSR for the large-scale upcycling of PET waste into novel high-value antibacterial materials (Fig. 7) [65]. Firstly, we have designed [C14][SIPA] with improved comprehensive properties comparing to [C16][SIPA]. Through subsequent reaction with ethylene glycol, an antibacterial modifier with hydroxyl–termination ([C14-EG]5) has been synthesized, enabling its application in PET modification. Utilizing the residual catalyst present in PET bottles, we facilitated the SSR of waste PET with [C14-EG]5, resulting in the production of high-value antibacterial PET. Notably, PET copolymer containing only 500 ppm of the antibacterial agent exhibited nearly 100% bactericidal activity against both gram-negative and gram-positive bacteria, demonstrating robust and enduring antibacterial efficacy even after 30 consecutive washing cycles. This work introduces a novel approach for the large-scale upcycling of waste PET into new high-value materials, offering potential for application under industry-relevant conditions.
Figure 7
Figure 7.
Development of a highly thermally stable monomer for the upcycling of PET waste, aimed at producing an antibacterial product through SSR. Copied with permission [65]. Copyright 2023, Wiley-VCH.
Second, we realize efficient bacterial infection combat via microenvironment-activated nanozyme-armed bacteriophages. Bacterial infection is one of the greatest challenges to public health, requiring new therapeutic methods [66–68]. Bacteriophage (abbreviated to phage) is an obligate predator of its bacterial host, which can recognize and hijack the host bacteria to generate progenies and ultimately lead to bacteria lysis [69]. Phage therapy has been proposed as an alternative to traditional antibiotics on the basis that phages only infect specific pathogens without destroying the normal microflora [70,71]. However, the bacterial populations at infection sites are usually a complex system, not only with multiple bacterial species, but also with a unique metabolic microenvironment, featuring low pH, high reactive oxygen species (ROS), specific enzymes, nutrient deficiencies, etc. [72]. Thus, a traditional phage therapy may be limited by narrow antibacterial spectrum and suffer from the harsh microenvironment, causing low bacterial eradication efficiency [73,74].
Herein, an innovative nanozyme-armed phage (phage@Pd) system is fabricated for combating bacterial infection (Fig. 8) [75]. The proposed phage@Pd preserves the function of the phages to achieve precise recognition and adhesion to the host Escherichia coli (E. coli). In aid of the phages, the ultrasmall palladium (Pd) nanozymes equipped with conspicuous pH-dependent peroxidase (POD)-like activity can generate toxic hydroxyl radical (•OH) around the bacteria in acidic and H2O2-overexpressed infection microenvironment while keeping inert in physiological conditions, thus realizing the noteworthy elimination of bacteria at infected site, in the meantime, ensuring the biological safety of phage@Pd in healthy tissues. In addition, the filamentous structure of phage@Pd can also enhance its bactericidal efficiency towards non-host bacteria by randomly tangling on them, indicating its possible broad-spectrum germicidal efficacy. Notably, phage@Pd can not only eradicate planktonic bacteria, but also kill the bacteria inside the biofilm in vitro. Both in vivo models of acute bacterial pneumonia or subcutaneous abscess, phage@Pd has shown significant activity in eliminating infection and promoting tissue recovery. These results demonstrate that the phage@Pd nanohybrid is a safe and effective antimicrobial agent, providing a new insight into development of advanced antibacterial materials.
Figure 8
Figure 8.
Schematic illustration of the structure of phage@Pd and its antimicrobial effect. Copied with permission [75]. Copyright 2023, Wiley-VCH.
Third, we deliver pH-sensitive nanomaterials for curcumin delivery. With the world stepping into a vicious cycle of antimicrobial resistance (AMR) [76–79], bacterial infections that were once considered treatable are now becoming untreatable and deadly [80,81]. In 2015, WHO put forward a global action plan, where countries were directed to individually develop their action plans to combat antibiotic resistance based on global strategy. As we are on the verge of a post-antibiotic era, it is critical time to re-vitalize the efficacy of antimicrobials or develop new non-antibiotic antibacterial strategies against resistant pathogens [82–87].
With the rapid development of nanotechnology, nanoparticles show great potential in the treatment of bacterial infections [88–90]. Supported by National Natural Science Foundation of China (Nos. 52293381, 52273154) and Natural Science Foundation of Zhejiang Province (No. LZ23B040002), we fabricated a series of antibacterial nanomaterials. About 65% of bacterial infections are related to biofilms. The bacterial cells in biofilms are 10 to 1000-fold more resistant to antibiotics than their planktonic counterparts. Biofilm microenvironment such as hypoxia and quorum sensing can greatly restrict the antibacterial effect of antibiotics [91]. In order to inhibit quorum sensing of biofilms, we designed an active targeted pH-sensitive nanoparticle (anti-CD54@Cur-DA NPs) for the delivery of quorum sensing inhibitor curcumin (Cur). Anti-CD54@Cur-DA NPs can respond to the acidic biofilm microenvironment and realize the change of nanoparticles’ size and charge, leading to enhanced biofilm penetration (Fig. 9). Compared to free Cur, Cur in the nanoparticles exhibited stronger capability in inhibiting quorum sensing, which could effectively down-regulate efflux pump-related genes and improve bactericidal performance of multiple antibiotics, including penicillin G, ciprofloxacin, and tobramycin [92].
Figure 9
Figure 9.
(A) The pH-responsiveness of anti-CD54@Cur-DA. (B) The delivery of Cur by anti-CD54@Cur-DA NPs to inhibit quorum sensing for enhanced antibiotic therapy against biofilm-associated infections. Copied with permission [92]. Copyright 2023, American Chemical Society.
Hypoxia is an important characteristic of biofilms. A “hypoxia-enhanced” strategy was adopted to improve the therapeutic efficacy of hypoxia-activated prodrug tirapazamine (TPZ). Aminated liposomes loaded with TPZ were prepared by thin film hydration method, and gold nanoparticles were grown by surface deposition to obtain gold nanozyme-hybrid liposomes (Lip@Au-TPZ). Gold nanozymes exhibit glucose oxidase-like properties that exacerbate hypoxic microenvironment of bacterial biofilm, activating hypoxia-activated prodrug TPZ to produce toxic radicals, showing excellent bactericidal properties [93]. Lip@Au-TPZ showed excellent efficacy on bacterial biofilm in mouse pneumonia model and mouse wound healing model. This strategy of exacerbating hypoxic bacterial biofilm microenvironment not only provides insight into the design of nanomaterials, but also brings new ideas for the selection of antibacterial drugs.
Besides antibiotics, we also developed innovative non-antibiotic antibacterial strategies. Transition metal ions are served as disinfectant thousand years ago. Although metal ions are widely used as bactericide in daily life, they are not used for in vivo antibacterial applications. We prepared Zn2+-gallic acid nanoflowers (ZGNFs) by a simple one-pot method without the addition of stabilizing agents [94]. Due to the strong interaction with the amino groups from teichoic acid of Gram-positive bacteria, ZGNFs could specifically ZGNFs adhere onto gram-positive bacteria high bactericidal effect toward various Gram-positive bacteria. More importantly, ZGNFs showed long-term retention in the infected corneal site in a MRSA-induced keratitis model, leading to remarkable antibacterial effect in vivo due to the self-targeting ability. Phototherapy, including photothermal therapy (PTT) and photodynamic therapy (PDT), is another important non-antibiotic antibacterial strategy due to its noninvasive intervention and satisfactory antibacterial efficiency. We rationally designed butterfly-shaped aggregation-induced emission luminogens (AIEgens) for imaging-assisted photothermal elimination of bacterial biofilms [95]. After AIEgens were encapsulated into cationic liposomes, the liposomes could eradicate various biofilms formed by Gram-positive bacteria and Gram-negative bacteria after 808 nm laser irradiation. Moreover, the NIR-Ⅱ AIE liposomes performed excellent antibacterial effect in both the P. aeruginosa biofilm-induced keratitis mouse model and methicillin-resistant Staphylococcus aureus (MSRA) biofilm-induced skin infection mouse model.
5.2
Biomedical materials
First, we design polymer cardiac patches for myocardial infarction treatment. Myocardial infarction is one of the leading causes of human deaths [96]. There is a high risk that the infarcted left ventricular (LV) myocardium turns into a fibrotic tissue, and pathologically remodels under cyclic stress exerted by adjacent myocardium, featuring LV dilation and cardiac wall thinning [97]. Epicardially implanted elastic patches could mechanically unload the infarcted myocardium and border zone, thus preserving cardiac functions and geometry by regulating the mechanosensing and cardiac contraction genes [98]. The concept of cardiac patch has been introduced for about 20 years, various patch designs have been demonstrated effective in animal models, including different polymer patches [98,99]. However, only one cardiac patch, CorPatch has been tested in clinically trials [100]. The bottleneck issue is the lack of a minimally invasive implantation method. In addition, incorporating synergetic function or bioactivities with mechanical support is desirable for obtaining improved therapeutic outcomes.
The dominant cardiac patch implantation technique is to suture the patch onto the epicardium in an open-chest surgery. To achieve minimally invasive implantation, sutureless patch fixation and the capability to transfer patches trough 10 mm-diameter channels (typical diameter of Trocars) are required. Inspired by the structural features of honeybee stingers, we fabricated stiff polymeric microneedles with unidirectionally backward-facing barbs, and embedded the microneedles into various elastomer films to produce self-interlocking microneedle patches [101]. The spirality of the barbing pattern was adjusted to reach an interlocking strength of 0.34 N per microneedle. In addition, the porous surface of the patch substrate was designed to absorb the micro-bleeding caused by microneedle puncturing and assist patch adhesion to the target tissue via coagulation. In rat myocardial infarction treatment, the microneedle patches firmly fixed on beating hearts, significantly reduced cardiac wall stress and strain in the infarcted myocardium, and maintained heart function and morphology [101]. We also successfully minimally invasively implanted 4.5 cm-diameter microneedle patch onto beating porcine heart in 10 min using thoracoscopy, free of sutures and adhesives [101]. Furthermore, we designed and synthesized a shape memory, self-healing polyurethane (PCLUSe) composed of semi-crystalline poly(ε-caprolactone) segments and interchangeable diselenide bonds. We delivered two folded PCLUSe through a 10 mm-diameter Trocar onto a beating canine heart, triggered shape recovery and self-assembly of the films into a larger single patch with laser irradiation in situ [102]. Hence, one can potentially combine the in situ shape recovery-assembly strategy and bioinspired fixation technique, and complete the minimally invasive patch implantation solution [102].
On the other hand, we developed a series of cardiac patches with therapeutic bioactivities additional to mechanical support. Last year, we reported multifunctional porous patches which promote cell ingrowth and tissue-patch integration, and scavenge reactive oxygen species [103,104]. However, these functions are not targeting at the cause of the pathological reactions, myocardial ischemia. This year, we developed a in situ photosynthesis system composed of intramyocardially injected chlorella and a biodegradable, flexible photonic patch (iCarP) for illumination on the chlorella to locally produce oxygen in the ischemic myocardium (Fig. 10) [105]. ICarP is featured with a micrometer thin air gap between a refractive polyester patch and the removable tapered optical fiber (TOF) embedded in the patch substrate. Epicardially adhered iCarP combines light diffraction by the TOF, dual refractions in the air gap, and reflection inside the polyester patch to obtain a bulb-like illumination, guiding light towards the myocardium [105]. We demonstrated that iCarP supports large area, high intensity, programmable illumination with deep penetration (> 1.5 cm). ICarP+/Light+ group had less apoptotic cardiac cells compared to iCarP+/Light− group. Consistent with alleviated tissue damage, photosynthesis treatment significantly increased LV ejection fraction compared to MI and iCarP+/Light− groups [105].
Figure 10
Figure 10.
The design, function and demonstrative application of the light scattering photonic device, illuminative cardiac patch (iCarP) for large area, deep illumination on internal organs and tissues. Copied with permission [105]. Copyright 2023, Springer Nature.
As illustrated above, in 2023, we made progress in minimally invasive implantation and functionalization of cardiac patches. Future studies will focus on safety and efficacy evaluation of cardiac patches in large animal models, to increase the clinical relevance of the developed technologies.
Second, we achieve drug coating of the vascular balloon. Percutaneous intervention through the drug-coated balloon (DCB) demonstrates the substantial potential for locally delivering anti-restenosis drugs to treat vascular lesions, without the presence of permanent implants [106–108]. However, clinically used anti-proliferative drugs, such as paclitaxel and rapamycin (RAPA), present cytotoxicity concerns and low efficiency in vivo [109,110]. In addition, the vascular restenosis and delayed re-endothelialization after balloon dilation disappoint the therapeutic effect of the DCB [111]. Therefore, the drug-coating design of the DCB is particularly important.
Vascular endothelial growth factor (VEGF) proficiently regulates the adhesion, aggregation, and proliferation of endothelial cells, fostering the development of neointima [112,113]. However, the relatively brief half-life of VEGF and its instability in the bloodstream may lead to poor therapeutic efficacy. In this context, we integrated plasmid DNA (pDNA) encoding VEGF and anti-proliferative medicine RAPA with protamine sulfate (PrS), a positively charged nuclear protein that markedly enhances the transfection efficiency of pDNA [114]. We found that the PrS/pDNA/RAPA nanocomposites coating displayed effective transfer capability from balloons to vessels both in vitro and in vivo, along with excellent stability and anticoagulant. Furthermore, we confirmed that the coating efficiently suppressed neointimal hyperplasia following balloon-induced vascular injuries by restraining the mammalian target of rapamycin (mTOR) signaling pathway and facilitated endothelial regeneration through an elevated expression of VEGF in vivo.
Excessive production of reactive oxygen species (ROS) at lesion sites typically leads to oxidative stress and heightened local inflammation, which results in tissue/cell damage, hindering endothelial functions, and exacerbating the process of intimal hyperplasia [115,116]. Therefore, the removal of ROS through antioxidants emerges as a prospective approach to modulate the pathological microenvironment of vessels. Boronic ester bonds, a type of reversible covalent bond, are renowned for their high sensitivity to ROS, and caffeic acid (CA) is found applied in the realms of anti-infectious, antioxidant, and anti-inflammatory attributed to its acrylic and catechol functional groups [117]. Here, a "ROS-responsive/scavenging prodrug" is introduced into balloon coating (Fig. 11) [118]. We incorporated the reversible phenylboronic ester-bearing CA prodrug with the endovascular delivery of microRNA-126 (miR126) as a combined strategy for pro-endothelial treatment. Our data suggest that CA and 4-hydroxybenzyl alcohol were released from the coating at high ROS level. The ROS-triggered release property of coating is essential for achieving potential on-demand and controllable delivery of drugs, particularly under inflammatory conditions marked by excessive ROS production. Meanwhile, the excessive ROS scavenging of coating helps maintain the activity and functions of miR126 for improved pro-endothelial therapy. With our drug delivery strategy, the ROS-responsive/scavenging prodrug/miRNA balloon coating efficiently mitigated local inflammation, impeded intimal hyperplasia, and facilitated endothelium recovery in a rat abdominal aorta restenosis model.
Figure 11
Figure 11.
Schematic illustration of vascular restenosis prevention by ROS-responsive/scavenging PBC@miR126-coated balloon. A reversible phenylboronic ester-bearing caffeic acid (CA) macromolecular prodrug (PBC) was designed for the controlled and on-demand dual-antioxidant release (CA and 4-hydroxybenzyl alcohol (HBA)) triggered by the local high ROS level, thereby exhibiting antioxidant and anti-inflammatory effects; the pro-endothelial microRNA-126 was introduced to further accelerate endothelialization. This endovascular ROS-responsive/scavenging prodrug/miRNA balloon coating efficiently inhibited long-term vascular restenosis. Copied with permission [118]. Copyright 2023, Wiley-VCH.
5.3
Biomacromolecule RNA labelling strategies and chemical modification functions
As a biomacromolecule, RNA plays key regulatory roles in almost every cellular process. In order to reveal diverse facets of RNA biology, RNA labelling has emerged as an indispensable tool for visualizing RNA locations, monitoring RNA dynamics and studying its interactions with other biomacromolecules [119,120]. In recent years, RNA metabolic labelling strategies have gained extensive attention, in which nucleoside analogues are incorporated into newly transcribed RNA through the nucleotide salvage pathway. We have developed a new nucleoside analogue, N4-allylcytidine (a4C), which is able to tag RNA through various pathways and can be further transformed into 3,N4-cyclized cytidine (cyc-C) in a catalyst-free, fast and complete manner (Fig. 12a) [121]. During RNA reverse transcription (RT), cyc-C induces base misincorporation and thus can be accurately identified by sequencing at single base resolution. With the chemical sequencing rationale of a4C, successful applications have been performed including pinpointing N4-methylcytidine methyltransferases’ substrate modification sites, metabolically labelling mammalian cellular RNAs, and mapping active cellular RNA polymerase locations.
Figure 12
Figure 12.
(a) Schematic diagram of RNA labelling applications based on a4C, a nucleoside analog with chemical sequencing power. Reproduced with permission [121]. Copyright 2024, Royal Society of Chemistry. (b) The workflow of a6A-seq, which takes advantage of a6A metabolic labelling on cellular mRNAs and profiles them in an immunoprecipitation-free and mutation-based manner. Reproduced with permission [122]. Copyright 2023, National Natural Science Foundation of China. (c) Schematic illustration to study the functions of NAT10 and mRNA ac4C modification in PSCs and during cell fate transitions. Reproduced with permission [123]. Copyright 2024, American Association for the Advancement of Science.
As RNA is highly dynamic in living organism, developing powerful tools to track RNA dynamic changes is of great significance. Recently, the integration of RNA metabolic labelling by nucleoside analogues with high-throughput RNA sequencing has been harnessed to study RNA dynamics. In our previous work, an adenosine analogue, N6-allyladenosine (a6A) was synthesized, and under mild iodination, it could be transformed into 1,N6-cyclized adenosine (cyc-A) to induce mismatch during RT. Based on this principle, we have developed a new method named a6A-seq (Fig. 12b) [122], which takes advantage of a6A metabolic labelling on cellular mRNAs and profiles them in an immunoprecipitation-free and mutation-based manner. We have successfully utilized a6A-seq to study cellular gene expression changes under a methionine-free stress condition. Compared with regular RNA-seq, a6A-seq could more sensitively detect the change of mRNA production over a time scale.
N4-Acetylcytidine (ac4C), has been identified as an epitranscriptomic RNA modification. N-Acetyltransferase 10 (NAT10) is the singular human enzyme to have both acetyltransferase and RNA binding activities, and more recently, NAT10 has been identified as the only known writer enzyme for ac4C modification on mRNAs. However, so far, the functions of NAT10 and mRNA ac4C modification in pluripotent stem cells (PSCs), or during cell fate transitions, remain largely unexplored. We have uncovered a previously unrecognized role for NAT10-ac4C in cell fate transitions, including reprogramming and differentiation (Fig. 12c) [123]. Mechanistically, we identified an important cross-talk between ac4C-mediated epitranscriptomic regulation and ANP32B-mediated chromatin signaling. These findings have provided a conceptual advance of cellular plasticity and can be harnessed for regenerative medicine.
6.
Photo-electro-magnetic functional polymers
First, we manipulate through-space conjugation in multiaryl alkanes to elucidate the clusteroluminescence (CL) mechanism and improve their performance. Through-space conjugation (TSC), involving the overlap and delocalization of electrons in two non-covalently bonded p orbitals [124,125], plays a pivotal role in CL [126,127], an abnormal luminescent phenomenon observed in non-conjugated structures at the aggregate state. Unlike the photophysical theory of through-bond conjugation (TBC) [128,129], which deals with the overlap and delocalization of electrons in two p orbitals connected by a covalent bond, TSC remains relatively understudied in terms of its structure-property relationship. Consequently, manipulating the strength of TBC remains challenging, leading to CL with short wavelengths and low efficiencies. Thus, the imperative task in CL research is to achieve controllable TSC.
To address this challenge, our research group has focused on manipulating TSC in nonconjugated multiaryl alkanes over the past year (2023). As depicted in Fig. 13a, we designed and synthesized seven tetraphenylalkanes (TPA) with varying alkane chain lengths (C1 to C7) [130]. Photophysical characterization of these molecules revealed an intriguing excited-state odd-even effect in the luminescence wavelength and efficiency. Notably, TPA molecules with an even number of carbon atoms in the alkane chains (C2-TPA, C4-TPA, and C6-TPA) exhibited strong visible emission with a quantum yield (QY) exceeding 43%. Conversely, those with an odd number of carbon atoms (C1-TPA, C3-TPA, and C5-TPA) showed negligible emission, with QY values below 8%. Through analysis of crystal structures and theoretical calculations, we attribute the quenched luminescence in C3-TPA and C5-TPA to intramolecular face-to-face packing of bilateral benzene rings, whereas bright emission in C2-TPA and C4-TPA results from intramolecular staggered packing. Additionally, we found that C1-TPA, with a crowded conformation, struggles to form intramolecular TSC, leading to quenched emission. Contrary to C1-TPA to C5-TPA, TSC in C6-TPA and C7-TPA with long alkane chains is regulated by intermolecular interactions. Crystal structure analysis revealed that luminescent C6-TPA exhibits intermolecular staggered packing akin to C2-TPA and C4-TPA, while non-emissive C7-TPA lacks well-defined crystallization due to weak intermolecular interactions. These findings underscore the critical role of subunit packing in determining TSC strength, with staggered packing favoring strong TSC. To validate this strategy, we introduced a methyl group to one side of C3-TPA, resulting in C3-Me-TPA, where face-to-face TSC is transformed into staggered TSC. As anticipated, emission in C3-Me-TPA was revived, with a QY of 42% [131].
Figure 13
Figure 13.
(a) Manipulation of the through-space conjugation (TSC) in tetraphenylalkanes. Reproduced with permission [130]. Copyright 2023, American Chemical Society. (b) Developing phenolic resin (PR) with red clusteroluminescence (CL) and the proposed emission mechanism. Reproduced with permission [134]. Copyright 2023, Wiley-VCH.
Building upon these findings [132,133], we modified nonconjugated phenolic resin (PR) by altering the monomers aldehyde (A) and phenol (B) (Fig. 13b) [134]. Introducing a methoxyl group increased electron density in the subunits, enhancing TSC, while additional phenyl rings also augmented TSC. Consequently, we observed red emission at 585 nm with a QY of 47% in An-MO-PR THF solution, with the solid-state An-MO-PR exhibiting near-infrared (NIR) CL at 680 nm. Theoretical calculations identified three emission species in PR derivatives: isolated phenol, through-space locally-excited state (TSLE), and through-space charge transfer state (TSCT).
In summary, manipulating TSC strength by altering subunit packing modes has been achieved, and NIR-CL PR has been successfully developed using these strategies. This not only sheds light on CL emission mechanisms but also enhances the application potential of CL luminophores by improving their performance.
Second, we realize room-temperature phosphorescence of hydrogels. Luminescent materials, especially those with room temperature phosphorescence (RTP) emission behaviors, have received increasing interests due to their versatile applications in biomedical and engineering fields [135–141]. As an important part, nontraditional polymeric phosphors with clusterization-triggered emission (CTE) properties attracted tremendous attention [127,142,143]. Such emission relies on the through-space conjugation of clustered nonconjugated electric rich groups (e.g., carbonyl group). This unique mechanism endows CTE-based polymeric phosphors with tunable RTP properties in solid state such as films or powders, which are environmentally sensitive and easy to be quenched in good solvent, making it challenging for the design and preparation of CTE-based RTP hydrogels [144–150]. This is because the hydration and swelling of gel matrix lead to cluster disaggregation and increased mobility of polymer chains, and thus enhanced nonradiative decays and reduced luminescence of the gels.
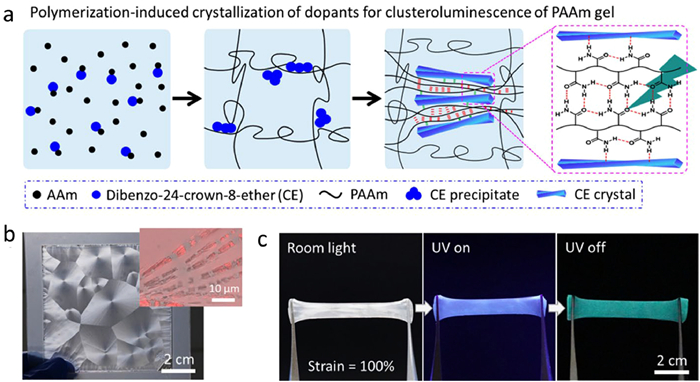
Recently, we proposed and demonstrated a facile strategy to develop RTP hydrogels by polymerization-induced crystallization of dopant molecules that results in segregation and confinement of gel matrix with carbonyl groups and thus CTE-based phosphorescence (Fig. 14a) [151]. Due to the differences in the solubilizing effect of acrylamide monomers and polyacrylamide (PAAm), dibenzo-24-crown-8-ether (CE) crystallized in situ and form large spherulite during the polymerization and gelation process to create crystal-doped polyacrylamide (CE-PAAm) hydrogels (Fig. 14b). The spherulite exhibited a discontinuous dendritic structure with the fragments of crystals composed of branched crystallite subunits with a length of 2–4 µm and a width of ~200 nm, which only restricted local deformation of the matrix and did not hamper large deformation of the intact gel. Therefore, the composite gels maintained good mechanical properties and achieved confinements of the carbonyl clusters between the polymer chains, resulting in RTP emission (Fig. 14c). With the 280 nm UV light irradiation, a typical CE-PAAm hydrogel exhibited green phosphorescence emission at 512 nm with a lifetime of 342 ms at room temperature. The excitation-wavelength dependence property and insensitivity to crown ether chelated ions indicated that carbonyl clusters were the dominant chromophores for the unique phosphorescence of CE-PAAm gel [150,152,153]. The formation and stabilization of clusters were attributed to the confinement of polymer chains between the crystallite subunits due to the steric resistant. The strategy showed its universality with various gel matrices and dopant molecules. The resultant gels exhibited versatile applications in the fields of chemical sensing, anti-counterfeiting, and soft actuators, which have been confirmed by several proof-of-concept examples and will benefit the design of robust hydrogel devices with phosphorescent behaviors.
Figure 14
Figure 14.
(a) Polymerization-induced crystallization of dopant molecules to form crystals that squeeze and confine polymeric matrix as cluster phosphors of the hydrogel. Digital photos of the dibenzo-24-crown-8-ether/polyacrylamide hydrogel in the as-prepared state (b) and after being stretched to a strain of 100% at room temperature under room light and after turning on and off 280 nm UV light (c). The inset in (b) is the confocal laser scanning microscope image showing the slender subcrystals of the spherulites in the composite gel. Copied with permission [151]. Copyright 2023, American Chemical Society.
Finally, we obtain long-range ordered bulk-heterojunctions (BHJs) by growing crystals in gels, and investigate its photodetecting performance. The long-range ordering of BHJs significantly enhances exciton diffusion and dissociation [154,155], as well as facilitating charge transport [156,157]. When combined with bi-continuous phase separation, efficient charge collection becomes possible, thereby yielding superior optoelectronic performance [158]. Ideally, single crystals are considered to be the most orderly-packed form, which is beneficial for longer exciton diffusion and faster charge transport. A feasible bio-inspired strategy to realize such a heterostructure involves crystallization within gel media, whereby the growing host crystals incorporate the neighboring guest materials from gel networks [159–162]. However, in some cases, the weak gel strength or an insufficient affinity between the crystal and the gel networks result in no gel incorporation [163,164]. While in other cases, only microcrystals assembled in the casted gels were obtained [165]. Recently, our group demonstrated poly[2–methoxy-5-(2-ethylhexyloxy)-1,4-phenylenvinylene] (MEH-PPV) gel incorporation inside C60 single crystals, and the resulting long-range ordered BHJs showed enhanced charge transfer and photodetection [166]. However, MEH-PPV suffers from relatively easier oxidizability, limited absorption range and poor crystallinity, in which carrier diffusion distance is less than 1 nm [167–169], spurring investigation towards crystalline gel networks. An alternative is poly(3-hexylthiophene) (P3HT). which provides hole diffusion length over 100 nm [170], and mobility exceeding 0.1 cm2 V−1 s−1 [171], forming gels comprised of crystalline fiber networks [172]. Achieving long-range ordered BHJs within a P3HT gel would enhance the overall structural ordering and, consequently, augment the optoelectronic properties. Nonetheless, the crystalline nature of P3HT gives a ~30 nm fiber width [173], leading to a larger barrier for the advancing fronts of growing crystal [174]. Thus, it remains to be elucidated whether such discrepancies would influence gel incorporation.
On the other hand, the emerging non-fullerene acceptors (NFAs) gain further attention due to merits such as broad and strong absorption, easier synthesis, relative stability. [175–181]. But unlike fullerene, NFAs typically exhibit a rigid planar conformation, with stronger π-π interaction [182]. Whether this densely-packed planar structure would alter gel incorporation behavior remains unexplored. Therefore, we attempted to grow C60 and (5Z,5′Z)-5,5′-((7,7′-(4,4,9,9-tetraoctyl-4,9-dihydro-s-indaceno[1,2-b: 5,6-b’]dithiophene-2,7-diyl)bis(benzo[c][1,2,5]thiadiazole-7,4-diyl))bis(methanylylidene))bis(3-ethyl-2-thioxothiazolidin-4-one) (O-IDTBR, a typical NFA) crystals inside the P3HT organogel [183]. As shown in Fig. 15a, C60 and O-IDTBR crystals were prepared by antisolvent interdiffusion into the P3HT gel. Crystals with lengths varying from tens to hundreds of microns were obtained within the gel. Afterwards, the gel incorporation was verified by etching. With slight etching, the penetration of P3HT fibrous networks was witnessed on the etched surfaces. The exposed fibers are ~30 nm in width, similar to the reported size (Fig. 15b) [173]. Subsequently, single-crystal X-ray diffraction (SC-XRD) reveals the crystallographic structures of gel-grown crystals, with an incident spot size of ~100 µm. Their diffraction patterns demonstrate a single set of symmetrically arranged spots (Fig. 15c), indicating the persistence of long-range ordering. Overall, our findings suggest that despite P3HT assembling into thick fibers, the fibrous networks could still be incorporated into both growing C60 and O-IDTBR single crystals while maintaining their long-range ordering.
Figure 15
Figure 15.
(a) Schematic illustration of the C60 and O-IDTBR crystallization in P3HT gel induced by anti-solvent. (b) SEM images of etched crystals. The exposed P3HT fibers are indicated by red arrows. Insert: an overall view of the etched crystal. (c) SC-XRD pattern of gel-grown C60 crystals viewed along the c-axis. (d) TRPL spectra of drop-casted P3HT gel, spin-coated (short-range ordered) BHJs, and gel-grown (long-range ordered) BHJs. (e) Photo/dark current under different illumination intensity, (f) bandwidth and (g) stability of photoresistors based on annealed blends (short-range ordered BHJs) and gel-grown single crystals (long-range ordered BHJs). Copied with permission [183]. Copyright 2023, Wiley-VCH.
Furthermore, we investigated the optoelectronic performance of such long-range ordered BHJs. Time-resolved photoluminescence (TRPL) spectra shows long-range ordered BHJs demonstrate a faster decay of 0.20 ns lifetime, accounting for 52% and 20% decrease in lifetime compared to pristine P3HT and short-range ordered BHJs, respectively (Fig. 15d). This is an indication of more efficient charge/energy transfer in long-range ordered BHJs. Apart from this, we tested long-range ordered BHJs as photodetectors. As shown in Figs. 15e-g, compared to short-range ordered BHJs, long-range ordered BHJs give 9.6 times and 154.9 times higher responsivities for C60: P3HT and O-IDTBR: P3HT BHJs, respectively. The cut-off frequency raised from 120 Hz to 550 Hz, indicating a faster respond time for long-range ordered BHJs. This could be attributed to the merits brought by long-range ordering: (1) Efficient charge/energy transfer; (2) augmented charge mobility; (3) higher charge collection efficiency due to the interpenetrating structure. Besides, long-range ordered BHJs demonstrated improved stability compared to the short-range ordered counterparts, with 2.1 and 26.4 times increase of time needed to decay to 80% of the initial photocurrent (T80) for C60: P3HT and O-IDTBR: P3HT BHJs, respectively. This is the result of two factors given by the single-crystalline nature: (1) Fewer defects protects the interiors from oxygen and water; (2) morphologically stable due to thermodynamic stability. In all, our demonstration showed the broad applicability of the bio-inspired strategy for constructing more efficient and versatile fullerene- and non-fullerene-based BHJs with long-range ordering, offering new perspectives for the development of advanced optoelectronic devices.
7.
Conclusion
In conclusion, aiming at the frontiers of polymer science advancements, the laboratory has made breakthroughs in its three main domains, including controlled polymerization for synthesizing novel functional polymers, elucidation of structure-property relationship by microstructure design, and high-performance polymer materials and devices fabrication. We briefly review the breakthroughs made by the laboratory in five research directions in 2023. First, in controllable catalytic polymerization, we invent a new silicon-centered organoboron binary catalyst, and develop a series of cooperative organocatalysts for ring-opening copolymerization. Second, in microstructure and rheology, we design axially encoded metafiber as multiple electronics, and construct interlayer entanglement for improved strength and toughness of artificial nacre materials. Third, in separating functional polymers, we monitor and regulate interfacial polymerization. Fourth, in biomedical functional polymers, we realize antibacterial materials including quaternary ammonium salt that enables polyethylene terephthalate recycling along with antibacterial function, nanozyme-armed phage for combating bacterial infection, and transition metal nanoparticles for antibacterial treatments. We also fabricate polymers for minimally invasive implantation and functionalization of cardiac patches, and ROS-responsive/scavenging prodrug/miRNA balloon coating for efficient drug delivery. As well we reveal methods and mechanisms for RNA labeling. Finally, in photo-electro-magnetic functional polymers, we manipulate through-space conjugation by altering subunit packing modes, synthesize room-temperature phosphorescent hydrogels by polymerization-induced crystallization of dopant molecules, and fabricate long-range ordered bulk heterojunctions in gel with further investigation of their performance as photodetectors. We are confident that these remarkable achievements and scientific perspectives will offer significant opportunities in the near future, advancing the synthesis, mechanism investigation, and practical application of versatile functional polymer materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
G. Yu, J. Ren, S. Yan, W. Yuan, H. Li, Small 19 (2023) e2302046. doi: 10.1002/smll.202302046
Figure 1
Schematic illustration about the key progresses of MOE Key Laboratory of Macromolecular Synthesis and Functionalization in 2023. Adapted with permission [5,44,48,105,118,121,122,130,151,183]. Copyright 2023, Wiley-VCH; Copyright 2023, Elsevier; Copyright 2023, American Chemical Society; Copyright 2023, Springer Nature; Copyright 2024, Royal Society of Chemistry; Copyright 2023, National Natural Science Foundation of China.
Figure 2
Silicon-centered binary (red) catalyst with higher activity than its bifunctional analogue (blue) for ring-opening polymerization of propylene oxide proposed by Wu et al. Reproduced with permission [5]. Copyright 2023, Wiley-VCH.
Figure 3
(a) ROCOP of PO and CO2 using BnBu3 combined with PtBu3. Reproduced with permission [21]. Copyright 2023, American Chemical Society. (b) ROCOP of TMO and CS2 using TEB combined with PPNCl. Reproduced with permission [25]. Copyright 2023, American Chemical Society. (c) ROCOP of COS, PO, and rac-lactide to yield stereo- and sequence-defined copolymers using various multifunctional organocatalysts. Reproduced with permission [29]. Copyright 2023, American Chemical Society.
Figure 4
(a) Designing concept of axially encoded metafiber from the code library. (b) Images of axially encoded metafibers. (c) Metafibers with axially functional asymmetry. Reproduced with permission [36]. Copyright 2023, Elsevier. (d) Schematic illustration of the localized strain engineering for highly sensitive sensor and IC protection. Reproduced with permission [37]. Copyright 2023, Wiley-VCH.
Figure 5
Structural model and overall properties of entangled nacre materials. (a) Failure models of entangled and regular nacre materials. Highly entangled polymer chains (crossing loops labelled in circles) strengthen energy dissipation to prevent catastrophic separation of sliding bricks than ordinary weak entangled case. (b) Photos of entangled nacre materials, including molecular entangled nacre fibers and films and nano entangled nacre papers. (c) The simultaneous increasing trend of toughness and strength of entangled nacres as Mw increases. Copied with permission [40]. Copyright 2023, American Chemical Society.
Figure 6
(a) Schematic illustration of the in situ and real-time monitoring of the interfacial polymerization based on the AIE effect. Reproduced with permission [42]. Copyright 2023, American Chemical Society. (b) Schematic of the effects of different alkane solvents on interfacial polymerization. Reproduced with permission [45]. Copyright 2023, Elsevier. (c) Schematic illustration of blade-coating-spraying-interfacial polymerization. Reproduced with permission [46]. Copyright 2023, Elsevier. (d) Schematic illustration of vacuum-assisted interfacial polymerization. Reproduced with permission [47]. Copyright 2023, Elsevier.
Figure 7
Development of a highly thermally stable monomer for the upcycling of PET waste, aimed at producing an antibacterial product through SSR. Copied with permission [65]. Copyright 2023, Wiley-VCH.
Figure 9
(A) The pH-responsiveness of anti-CD54@Cur-DA. (B) The delivery of Cur by anti-CD54@Cur-DA NPs to inhibit quorum sensing for enhanced antibiotic therapy against biofilm-associated infections. Copied with permission [92]. Copyright 2023, American Chemical Society.
Figure 10
The design, function and demonstrative application of the light scattering photonic device, illuminative cardiac patch (iCarP) for large area, deep illumination on internal organs and tissues. Copied with permission [105]. Copyright 2023, Springer Nature.
Figure 11
Schematic illustration of vascular restenosis prevention by ROS-responsive/scavenging PBC@miR126-coated balloon. A reversible phenylboronic ester-bearing caffeic acid (CA) macromolecular prodrug (PBC) was designed for the controlled and on-demand dual-antioxidant release (CA and 4-hydroxybenzyl alcohol (HBA)) triggered by the local high ROS level, thereby exhibiting antioxidant and anti-inflammatory effects; the pro-endothelial microRNA-126 was introduced to further accelerate endothelialization. This endovascular ROS-responsive/scavenging prodrug/miRNA balloon coating efficiently inhibited long-term vascular restenosis. Copied with permission [118]. Copyright 2023, Wiley-VCH.
Figure 12
(a) Schematic diagram of RNA labelling applications based on a4C, a nucleoside analog with chemical sequencing power. Reproduced with permission [121]. Copyright 2024, Royal Society of Chemistry. (b) The workflow of a6A-seq, which takes advantage of a6A metabolic labelling on cellular mRNAs and profiles them in an immunoprecipitation-free and mutation-based manner. Reproduced with permission [122]. Copyright 2023, National Natural Science Foundation of China. (c) Schematic illustration to study the functions of NAT10 and mRNA ac4C modification in PSCs and during cell fate transitions. Reproduced with permission [123]. Copyright 2024, American Association for the Advancement of Science.
Figure 13
(a) Manipulation of the through-space conjugation (TSC) in tetraphenylalkanes. Reproduced with permission [130]. Copyright 2023, American Chemical Society. (b) Developing phenolic resin (PR) with red clusteroluminescence (CL) and the proposed emission mechanism. Reproduced with permission [134]. Copyright 2023, Wiley-VCH.
Figure 14
(a) Polymerization-induced crystallization of dopant molecules to form crystals that squeeze and confine polymeric matrix as cluster phosphors of the hydrogel. Digital photos of the dibenzo-24-crown-8-ether/polyacrylamide hydrogel in the as-prepared state (b) and after being stretched to a strain of 100% at room temperature under room light and after turning on and off 280 nm UV light (c). The inset in (b) is the confocal laser scanning microscope image showing the slender subcrystals of the spherulites in the composite gel. Copied with permission [151]. Copyright 2023, American Chemical Society.
Figure 15
(a) Schematic illustration of the C60 and O-IDTBR crystallization in P3HT gel induced by anti-solvent. (b) SEM images of etched crystals. The exposed P3HT fibers are indicated by red arrows. Insert: an overall view of the etched crystal. (c) SC-XRD pattern of gel-grown C60 crystals viewed along the c-axis. (d) TRPL spectra of drop-casted P3HT gel, spin-coated (short-range ordered) BHJs, and gel-grown (long-range ordered) BHJs. (e) Photo/dark current under different illumination intensity, (f) bandwidth and (g) stability of photoresistors based on annealed blends (short-range ordered BHJs) and gel-grown single crystals (long-range ordered BHJs). Copied with permission [183]. Copyright 2023, Wiley-VCH.


 DownLoad:
DownLoad:













 下载:
下载:
















